

Family: Herelleviridae
Jakub Barylski, Andrew M. Kropinski, Nabil-Fareed Alikhan, Małgorzata Łobocka and Evelien M. Adriaenssens
Corresponding author: Jakub Barylski ([email protected])
Edited by: Evelien M. Adriaenssens
Posted: July 2020, updated September 2020, September 2022
PDF: ICTV_Herelleviridae.pdf (2020 version)
Summary
Members of the family Herelleviridae are bacterial viruses infecting members of the phylum Firmicutes (Table 1 Herelleviridae). In the Ninth Report of the ICTV, this group was reported as the genus SPO1-like viruses (https://ictv.global/taxonomy/taxondetails?taxnode_id=202100268). The virions have myovirus morphology, i.e., a head-tail structure with a long, contractile tail, and an icosahedral head. Genomes are dsDNA of 125–170 kbp (Table 1 Herelleviridae).
Table 1 Herelleviridae. Characteristics of members of the family Herelleviridae
Characteristic | Description |
Example | Bacillus phage SPO1 (FJ230960), species Okubovirus SPO1 genus Okubovirus |
Virion | Head-tail morphology with contractile tail, heads generally isometric with diameters of 85–100 nm showing capsomers, uncontracted tails of 130–185 nm in length |
Genome | Linear, terminally redundant, non-permuted dsDNA of 106–170 kbp |
Replication | Phage-encoded DNA polymerase |
Translation | Bacterial translation |
Host range | Bacteria of the phylum Firmicutes |
Taxonomy | Realm Duplodnaviria, kingdom Heunggongvirae, phylum Uroviricota, class Caudoviricetes, 5 subfamilies, 34 genera and 119 species |
Virion
Morphology
Virions have isometric, icosahedral heads of 85–100 nm in diameter. The heads show clear capsomers, i.e., the subunits of the capsid are arranged in pentons and hexons that are assembled into the isometric, icosahedral capsid. The uncontracted tails are 130–185 nm in length. The tails have a baseplate of approximately 60 nm and a small collar (Figure 1.Herelleviridae).
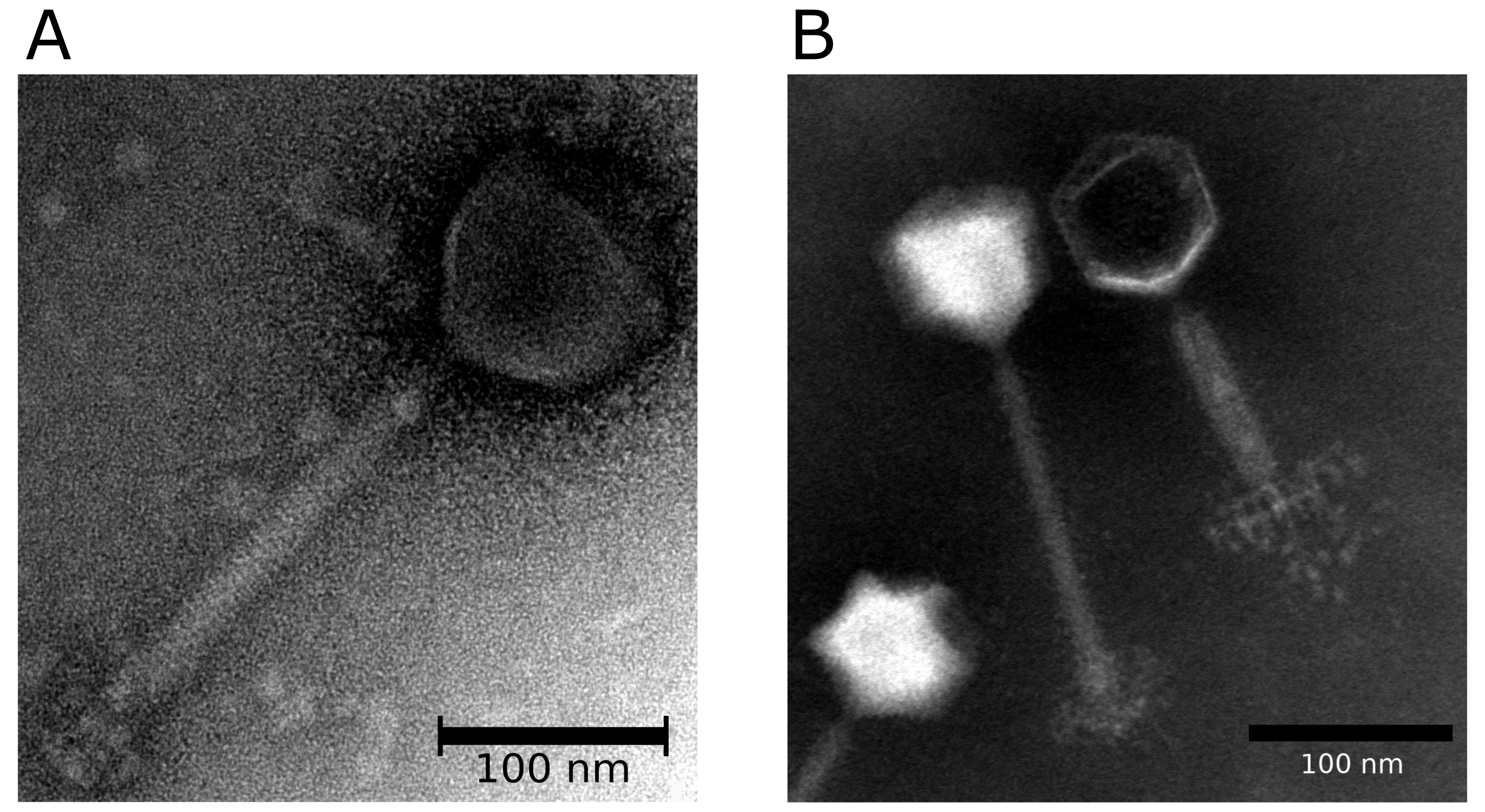 |
| Figure 1.Herelleviridae. Morphology of members of the family Herelleviridae. (A) Transmission electron micrograph of Bacillus phage phiAGATE, a member of the subfamily Bastillevirinae. Virions in this sample were concentrated from bacteria-free lysates and stained with 2% uranyl acetate. (B) Transmission electron micrograph of Staphylococcus phage 676Z, a member of the subfamily Twortvirinae. Particles from this sample were concentrated from bacteria-free lysate, washed twice with 0.1 M ammonium acetate (pH 7.2) applied to a formvar carbon coated copper grid and stained with 2% sodium phosphotungstate. Observations were performed in the Laboratory of Electron Microscopy at the Nencki Institute of Experimental Biology, Warsaw, Poland by Monika Hejnowicz, IBB, Polish Academy of Sciences, Warsaw. Scale bar represents 250 nm. In both (A) and (B) stained grids were examined in a JEM 1400 (JEOL Co., Japan, 2008) transmission electron microscope. |
Physicochemical and physical properties
Bacillus phage SPO1 buoyant density in CsCl is 1.54 g/cm3.
Nucleic acid
The genomes of herelleviruses consist of linear dsDNA with long terminal repeats of 2–16 kbp (Perkus and Shub 1985, Klumpp et al., 2008, Łobocka et al., 2012, Cadungog et al., 2015). Genomes are of 106–170 kbp (based on NCBI records with genomes of known members of the family). tRNAs are encoded by some members. The genome of Bacillus phage SPO1 has thymidine replaced with 5-hydroxymethyluridine. DNA modifications are also present in some members of the family (Okubo et al., 1972, Parker and Eiserling 1983, Klumpp et al., 2010).
Proteins
The virion of Bacillus phage SPO1 comprises at least 35 proteins as judged from electropherograms of purified particles (Parker and Eiserling 1983).
Lipids
No lipids reported.
Carbohydrates
No carbohydrates reported.
Genome organization and replication
The genomes of herelleviruses are linear dsDNA of 106–170 kbp, typically with long terminal repeats of various lengths (3.1–20 kbp). Genomes encode 126–301 proteins, and 0–24 tRNAs. For Bacillus phage SPO1, the majority of the coding sequences are in the same orientation; two islands encoding hypothetical proteins are transcribed from the opposite strand (Figure 2.Herelleviridae). The terminal repeats of SPO1 contain a host-takeover module involved in early stages of phage propagation. Host DNA and RNA synthesis is shut off in first minutes of infection. Phage DNA replication depends on phage-encoded DNA polymerase, which is similar to bacterial DNA polymerases. Transcription is mediated by host RNA polymerase which binds phage-encoded sigma factors to turn on the programmed cascade of phage gene expression (Stewart et al., 2009). The genes are categorized as early, middle and late, according to the profiles of their transcription in time (Stewart et al., 2009). Introns have been identified in a number of herellevirus genomes (Goodrich-Blair et al., 1990, Lavigne and Vandersteegen 2013).
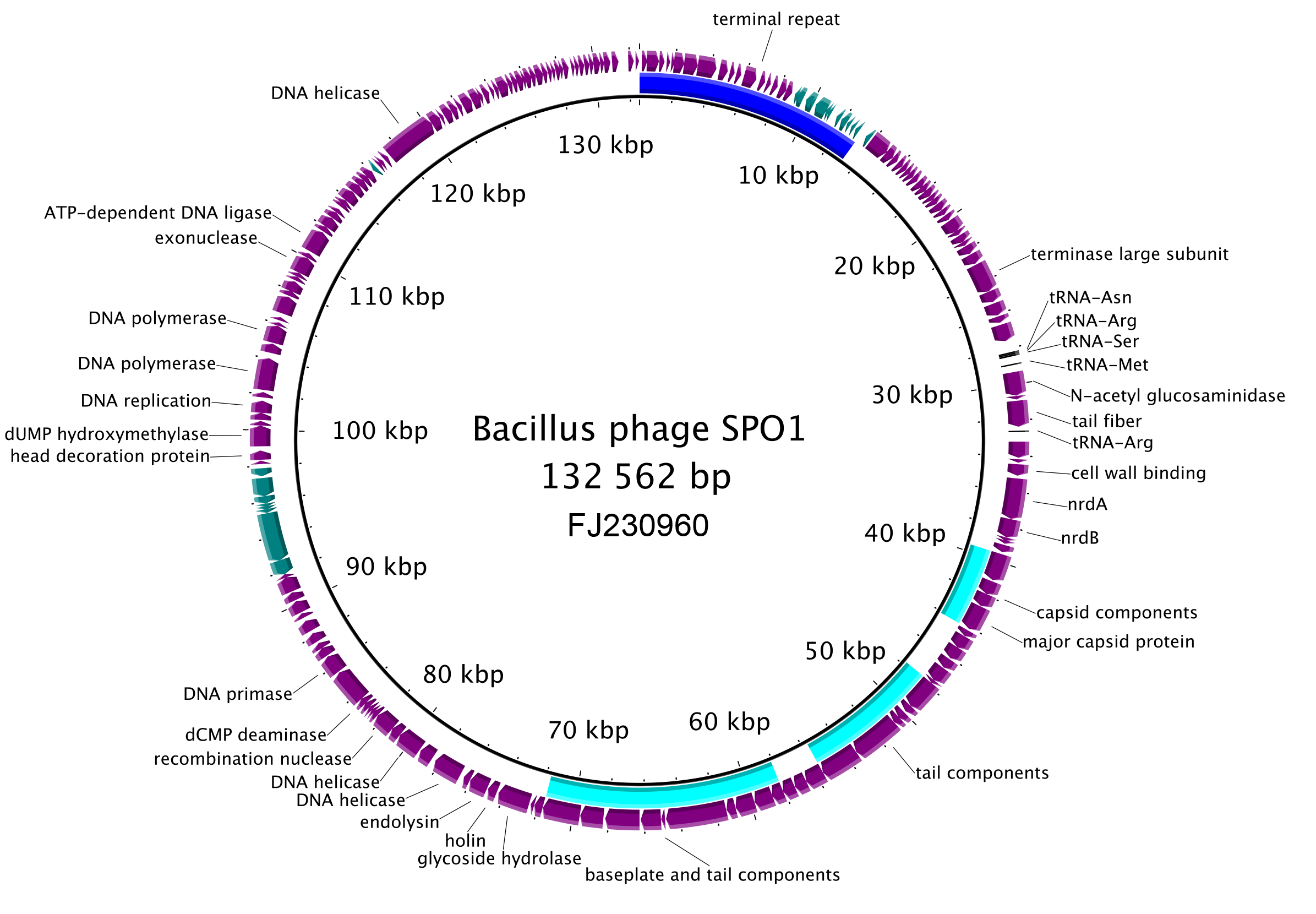 |
| Figure 2. Herelleviridae. Genome organisation of Bacillus phage SPO1, a typical member of the family Herelleviridae. The terminal repeat area is indicated with a blue arc, structural modules with a cyan arcs. The predicted coding sequences on the plus-strand are in purple, those on the minus-strand in teal. The five predicted tRNAs are indicated in black. This figure was generated with BRIG (Alikhan et al., 2011). |
Table 2.Herelleviridae. Core genes with predicted functions shared among all members of the family*
| Gene | Prokaryotic virus orthologous group number |
| DnaB-like helicase | VOG0025, VOG4691 |
| baseplate J-like protein | VOG4644 |
| tail sheath protein | VOG0067 |
| terminase large subunit (intron-invaded) | VOG0051 |
| major capsid protein | VOG0061 |
| prohead protease | VOG4568 |
| portal protein | VOG4556 |
| DNA primase | VOG4551 |
| DNA polymerase I | VOG0668 |
| RNA polymerase | VOG0118 |
| recombination exonuclease | VOG4575 |
| recombination endonuclease | VOG0083 |
| tail tape measure protein | VOG0069 |
| tail tube protein | VOG0068 |
* at a 50% amino acid sequence identity threshold at minimum 50% gene coverage or by prokaryotic Virus Orthologous Group (pVOG) analysis (Barylski et al., 2020)
Biology
The phages belonging to this family are reported to be obligately lytic, but some may be able to cause persistent infection, pseudolysogenic infection or a carrier state (Schuch and Fischetti 2009, Yuan et al., 2015). Herelleviruses infect bacteria belonging to the phylum Firmicutes and have a worldwide distribution.
Derivation of names
Agatevirus: from Bacillus phage phiAGATE, the exemplar isolate of the species Agatevirus agate.
Bastillevirinae, Bastillevirus: from Bacillus phage Bastille
Bequatrovirus: from Bacillus phage B4, the exemplar isolate of the species Bequatrovirus B4 (B [Be] followed by quatro, derived from Italian for four, quattro).
Baoshanvirus: derived from the place of origin (the Chinese district Baoshan) of Staphylococcus phage phiSA_BS2, the exemplar isolate of the species Baoshanvirus BS2,
Brockvirinae: named in honour of Thomas D. Brock (1926–2021), the eminent American microbiologist and educator known for his discovery of hyperthermophiles, who worked on Streptococcus phages early in his career.
Caeruleovirus: derived by association from Bacillus phage Deep Blue, the exemplar isolate of the species Caeruleovirus deepblue, caerulean being a shade of blue.
Eldridgevirus: derived from Bacillus phage Eldridge - the exemplar isolate of the species Eldridgevirus eldridge.
Elpedvirus: derived phonetically from Lactobacillus phage LpeD, the exemplar isolate of the species Elpedvirus LpeD.
Goettingenvirus: named after the city of Göttingen, the location of the Genomic and Applied Microbiology & Genomics Laboratory, Georg-August-University,where Bacillus phage vB_BmeM-Goe8), the exemplar isolate of the species Goettingenvirus goe8 was isolated.
Grisebachstrassevirus: named after a street in Göttingen (Grisebachstraße), the location of the Genomic and Applied Microbiology & Genomics Laboratory, Georg-August-University, where the Bacillus phage vB_BsuM-Goe3, the exemplar isolate of the species Grisebachstrassevirus goe3 was isolated.
Harbinvirus: derived from Northeast Agricultural University in Harbin, China, where Lactobacillus phage Lpa804 the exemplar isolate of the representative species Harbinvirus Lpa804, was isolated.
Herelleviridae named in honour of the 100th anniversary of the discovery of bacteriophages by Félix d’Hérelle in 1917.
Hopescreekvirus: derived from Hopes Creek Road - the location of Ecolyse Inc. Labs where Lactobacillus phage LfeInf, the exemplar isolate of the species Hopescreekvirus LfeInf was isolated.
Jasinkavirinae: named in honour of Stanisława Jasińska-Lewandowska (1921–1998), a Polish scientist who was one of the first to study species of Listeria and their viruses (note that due to an error in the original taxonomic proposal, the subfamily name currently lacks the second "s" from Jasińska).
Jeonjuvirus: named after city of Jeonju in western South Korea, the location of the Department of Food Science and Technology of Chonbuk National University where Bacillus phage BSP38, the exemplar isolate of the species Jeonjuvirus BSP38 was first isolated.
Kayvirus: derived phonetically from the name of Staphylococcus phage K the exemplar isolate of the species Kayvirus kay.
Kochikohdavirus: derived from Kohda, Kochi City, Japan, where Enterococcus phage phiEF24C, the exemplar isolate of the species Kochikohdavirus EF24C, was isolated.
Matervirus: derived from Bacillus phage Mater, the exemplar isolate of the species Matervirus mater.
Moonbeamvirus: derived from Bacillus phage Moonbeam , the exemplar isolate of the species Moonbeamvirus moonbeam.
Mooreparkvirus: derived from Moorepark Food Research Centre, Cork, Republic of Ireland where Lactobacillus phage Lb338-1, the exemplar isolate of Mooreparkvirus Lb3381, was isolated.
Nitunavirus: derived phonetically from Bacillus phage phiNIT1, the exemplar isolate of the species Nitunavirus NIT1 (i.e., nit followed by una meaning one).
Okubovirus: named in honour of Shunzo Okubo (1930–1978), former Professor of Genetics at Osaka University who isolated Bacillus phage SPO1 in 1964 and carried out the original studies on this virus, now the exemplar isolate of the species Okubovirus SPO1.
Pecentumvirus: derived phonetically from the name of the isolate, Listeria phage P100 ("pe" followed by centum meaning 100 in Latin).
Salchichonvirus: derived from Salchichón – the name of a Spanish summer sausage and the source of Lactobacillus phage LP65, the exemplar isolate of the species Salchichonvirus LP65, and the first known member of the genus.
Schiekvirus: named after the German virologist Wolfgang Schiek (Hygiene-Institut, University of Göttingen) who was one of the first scientists to work on Enterococcus phages. Sciuriunavirus: derived from the species epithet of the host (Staphylococcus sciuri) and last part of the name of the isolate Staphylococcus phage vB_SscM-1, the exemplar isolate of the species Sciuriunavirus SscM1, and the first sequenced phage of Staphylococcus sciuri (Jurczak-Kurek et al., 2016).
Sepunavirus: derived from the name of the isolate Staphylococcus phage phiIBB-SEP1 (i.e., sep followed by una, meaning one).
Shalavirus: named after Lake Shala, the place of isolation of Bacillus phage Shbh1, its first known member.
Silviavirus: indirectly derived from Staphylococcus phage Remus, the exemplar isolate of the species Silviavirus remus. In Roman mythology, Silvia was the mother of Romulus and Remus, after which two phage strains classified in this genus were named.
Siminovitchvirus: named in honour of Louis Siminovitch (1920–2021), a Canadian molecular biologist who studied phages early in his academic career.
Siophivirus: derived from Bacillus phage SIOphi, the first known member of the genus.
Spounavirinae derived from Bacillus phage SPO1 (i.e., spo and una, meaning one), the first recognised isolate of this subfamily.
Tsarbombavirus: derived from Bacillus phage TsarBomba, the exemplar isolate of the species Tsarbombavirus tsarbomba.
Twortvirinae: named in honour of Frederick William Twort (1877–1950), the English bacteriologist who discovered prokaryotic viruses in 1915.
Twortvirus: derived from the name of Staphylococcus phage Twort, the exemplar isolate of the species Twortvirus twort, the isolate itself named after Frederick William Twort (1877–1950), the English bacteriologist who discovered prokaryotic viruses in 1915.
Tybeckvirus: named in honour of the Finnish scientist Ethel Ingeborg Tybeck (1923–1982) who was one of the first people to isolate phages infecting Lactobacillus sp.
Watanabevirus: named after the pioneering Lactobacillus phage scientist, Dr. Kenji Watanabe.
Wphvirus: derived from Bacillus phage W.Ph., the exemplar isolate of the species Wphvirus WPh.
Subfamily demarcation criteria
Subfamilies (Table 3.Herelleviridae) are identified as well-supported monophyletic groups based on phylogenetic analysis of concatenated core gene markers and single core genes (Figure 3.Herelleviridae). Within a subfamily, members are 20–25% identical in translated nucleotide content as calculated with the tBLASTx algorithm. Nucleotide sequence identity between members of different subfamilies is hard to detect using BLAST-based methods, there being only small patches of similar sequence, usually amounting to less than 1% of the genome length.
Table 3.Herelleviridae. The subfamilies of Herelleviridae
Subfamily | Genera | Host | Genome (kbp) | Terminal repeats* |
Bastillevirinae | 15 | Bacillus sp., Priestia sp. | 138–167 | Long |
Brockvirinae | 2 | Enterococcus sp. | 140–155 | ND |
Jasinkavirinae | 1 | Listeria monocytogenes | 131–138 | ND |
Spounavirinae | 2 | Bacillus sp. | 132–146 | Long |
Twortvirinae | 6 | Staphylococcus | 127–150 | Long |
*ND Not Determined for all members
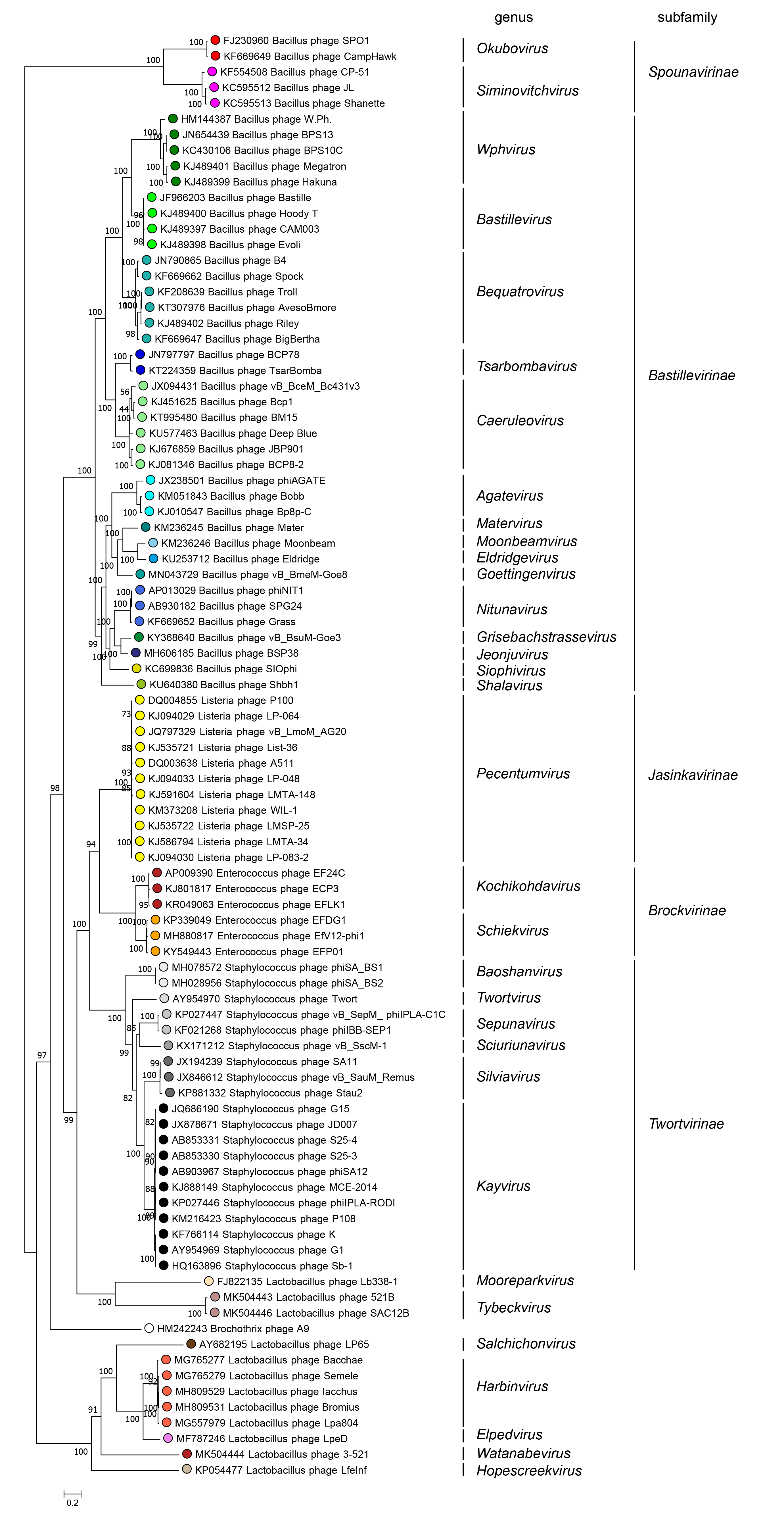 |
| Figure 3.Herelleviridae. All genomes were re-annotated using Prokka then orthologous protein clusters were constructed using the GET_HOMOLOGUES software, using COGtriangles algorithm (Kristensen et al., 2010) was applied with a 50% sequence identity threshold, 50% coverage threshold, and an e-value cutoff equal to 1×10-5. After a manual curation we selected 12 consistently-predicted protein groups (encoded by genes with well-defined boundaries and without introns) present in all analyzed genomes for further analysis. The members of these clusters were aligned using Clustal Omega with default parameters (Sievers et al., 2011). The resulting alignments were concatenated and analyzed with the IQ-TREE pipeline (Nguyen et al., 2015, Chernomor et al., 2016, Kalyaanamoorthy et al., 2017, Hoang et al., 2018) to generate the approximate "species tree" based on the partitioned evolution model. Analysis was run in 100 replicates to select the final tree with best log-likelihood score. Ultrafast boostraps (UFBOOT) scores out of 100 indicate branch support. Coloured circles indicate members of the same genus, while white circles are asssigned to a speces but not a genus. This phylogenetic tree and corresponding sequence alignment are available to download from the Resources page. |
Genera not assigned to subfamilies
There are currently eight genera not assigned to a subfamily. With the exception of members of the species Klumppvirus A9 (the only species in the genus Klumppvirus) which infect bacteria of the genus Brochothrix, these are all Lactobacillus and Lacticaseibacillus-infecting phages that will probably be grouped into higher-ranking taxa after the discovery of more similar sequences.
Genus demarcation criteria
Genera are well-supported monophyletic clades in genome-based phylogenies and in (concatenated) marker gene phylogenies. Members of a genus share at least 60% nucleotide identity across the genome. The genome organisation is conserved. Members of a genus generally infect members of the same bacterial genus.
Species demarcation criteria
The species demarcation criteria are the same for all species within the family. Members of the same species are more than 95% identical in genome nucleotide sequence, including the terminal repeat region. Phages with genomes that differ in more than 5% are assigned to different species.
Relationships within the family
Phylogenetic analysis of 10 core gene products shared among all herelleviruses (Barylski et al., 2020)has identified five subfamilies, 34 genera and 92 species. Eight genera have not been assigned to a subfamily (Figure 3.Herelleviridae). Phylogenetic relationships reconstructed by most single gene phylogenies are largely consistent, although shuffling between subfamilies has been observed for the tail tube protein (Barylski et al., 2020).
Relationships with other taxa
Herelleviruses share morphological similarity with other myoviruses, i.e. members of the subfamily Tevenvirinae (family Straboviridae) with the exception that herellevirus heads are not prolate, unlike heads of tevenvirins.