

Family: Geminiviridae
Genus: Eragrovirus
Distinguishing features
Eragroviruses have an unspliced replication-associated protein (Rep) and a unique genome organization, with a 5′-TAAGATTCC-3′ virion strand origin of replication (like becurtoviruses). Their genomes resemble a chimera with elements taken from isolates of different geminivirus genera. The C2 ORF is a positional analog of begomovirus, topocuvirus, and curtovirus TrAP/TrAP-like genes. The V1 ORF (which encodes the geminivirus capsid protein) is most similar to those of Eurasian mastreviruses. The eragrovirus Rep appears most closely related to those of begomoviruses, curtoviruses, and topocuviruses.
Virion
See discussion under family description.
Genome organization and replication
The arrangement of potentially-expressed open reading frames (ORFs) within eragrovirus genomes is similar to those described for other geminiviruses (Figure 1. Eragrovirus). The locations of the two largest virion sense ORFs (V1 and V2, respectively) correspond to the positions of cp and mp genes found in other geminiviruses with monopartite genomes. Similarly, the two largest complementary sense ORFs (C1 and C2) occur in the same positions as rep and trap/trap-like genes found in topocuviruses, begomoviruses and curtoviruses. Whereas the predicted expression products of the V1 and C1 ORFs share identifiable similarities with geminivirus coat protein (CP) and Rep proteins, respectively, the other ORFs have no known homologues. For example, whereas the V2 ORF is in an analogous position to the mp genes of other geminiviruses, its translated sequence lacks the large hydrophobic domain that characterizes mastrevirus movement proteins (MP) (Varsani et al., 2014b).
|
|
|
Figure 1. Eragrovirus. Genomic organization of eragroviruses. ORFs are denoted as being encoded on the virion-sense (V) or complementary-sense (C) strand. The positions of the stem-loop motif containing the conserved 5′-TAAGATTCC-3′ sequence in the intergenic region 1 (IR-1) and the location of intergenic region 2 (IR-2) are shown. |
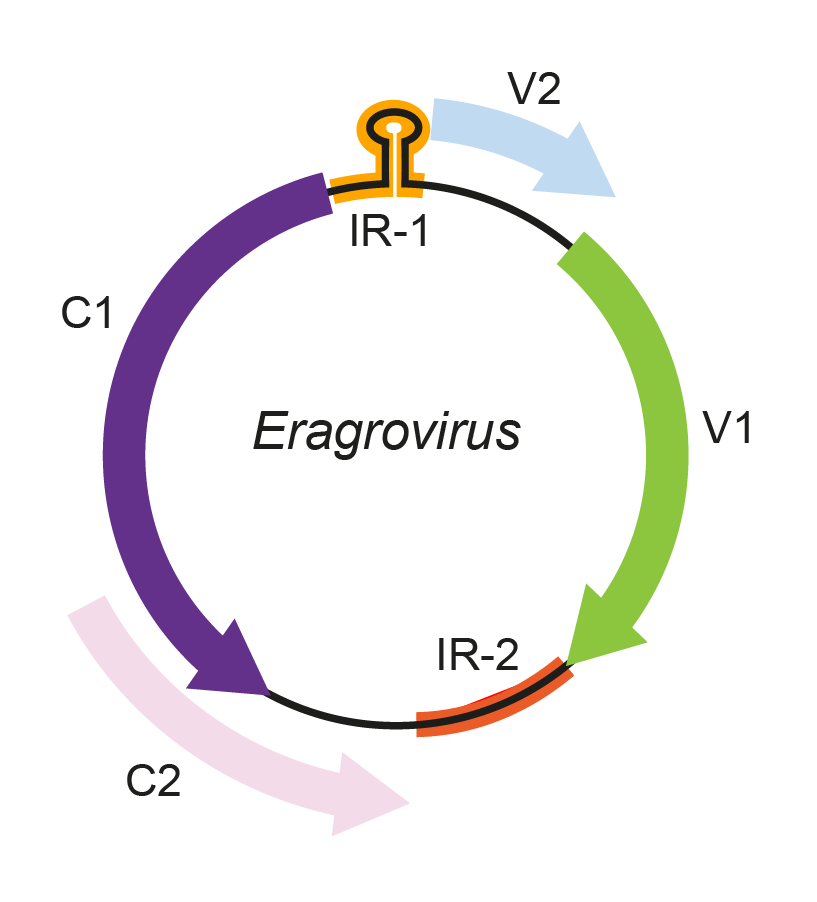
As with mastreviruses and becurtoviruses, the eragrovirus genome contains two intergenic regions (IR). However, in the case of eragroviruses, the analogue of the mastrevirus long IR (location of the v-ori and transcription start sites) is smaller than the analogue of the mastrevirus short IR (location of the complementary strand replication origin and transcription termination sites). The v-ori-containing IR is referred to as IR-1, and the larger IR as IR-2. A number of sequence elements are present within IR-1 and IR-2 that, by analogy with other geminiviruses, are potentially involved in replication and/or transcription. As with becurtoviruses, eragroviruses have a 5′-TAAGATTCC-3′ sequence rather than the usual 5′-TAATATTAC-3′ sequence found in the origin of replication of almost all other geminiviruses. Besides this similarity with becurtoviruses, the overall structural arrangement of IR-1 is most similar to the IRs of begomoviruses, curtoviruses and topocuviruses. Directly repeated sequences in IR-1 between the probable rep initiation codon and a potential complementary-sense transcript TATA box, resemble the arrangement of begomovirus, topocuvirus and curtovirus iterated sequence elements implicated in v-ori recognition and binding by Rep (Varsani et al., 2009).
Biology
Host range
All known isolates of the only species in the genus, Eragrostis curvula streak virus, have been isolated from the monocotyledonous species Eragrostis curvula (weeping love grass, a perennial grass with a distribution from southern to East Africa) in the Kwa-Zulu Natal region of South Africa. The virus causes streak symptoms similar to those encountered in mastrevirus-infected grasses (Varsani et al., 2009).
Species demarcation criteria
As the genus includes a single species, no species demarcation criteria have been defined. Six isolates of Eragrostis curvula streak virus (ECSV) all share >91.6% genome-wide sequence identity and, based on the distribution of pairwise identities, the Geminiviridae Study Group propose a tentative strain demarcation threshold of 94% (Varsani et al., 2014b). Thus, pairs of eragrovirus full-genome sequences that have >94% pairwise identity should be considered as members/variants of the same strain.