

Family: Geminiviridae
Genus: Citlodavirus
Distinguishing features
The most distinctive feature of members of the genus Citlodavirus is that their genomes are between 12% and 30% larger than the genomes of other known monopartite geminiviruses. This is primarily attributable to the length of their presumed mp gene (891–921 nt) which is up to three times larger than that of monopartite geminiviruses but is approximately the same length as that of bipartite begomoviruses. However, in bipartite begomoviruses the 714 to 1107 nt mp gene that encodes a protein with detectable identity to the movement proteins of citlodaviruses is found on the DNA-B molecule. This suggests that citlodavirus members may represent an intermediary step in the evolution of bipartite begomoviruses (with genomes of approximately 5.3 kb) from monopartite geminiviruses (with genomes that are generally between 2.7 and 3 kb). All citlodaviruses are monopartite and their genomes contain the 5′-TAATATTAC-3′ nonanucleotide at the v-ori (Fontenele et al., 2018).
Virion
See discussion under family description.
Genome organization and replication
The citlodavirus genomic arrangement contains a putative stem-loop structure which includes the nonanucleotide motif 5′-TAATATTAC-3′, highly conserved at the origin of virion strand replication in geminivirus genomes. The genome contains six open reading frames. The complementary strand of the genome potentially encodes geminivirus-like RepA and/or Rep proteins. Rep and RepA proteins are expressed from an alternatively spliced complementary strand transcript, as it was identified by small RNA (sRNA)-seq and RNA-seq reads mapping of paper mulberry leaf curl virus 2 antisense transcripts (Qiu et al., 2020) .
The virion sense genome strand potentially encodes a coat protein, a movement protein, and two other small hypothetical proteins, V2 and V3 (Fontenele et al., 2018) (Figure 1. Citlodavirus).
|
|
|
Figure 1. Citlodavirus. Genomic organization of citlodaviruses. ORFs are denoted as being encoded on the virion-sense (V) or complementary-sense (C) strand. The positions of the stem-loop motif containing the conserved 5′-TAATATTAC-3′ sequence in the long intergenic region (LIR). Intron is predicted to occur between ORFs C1 and C2. SIR, short intergenic region |
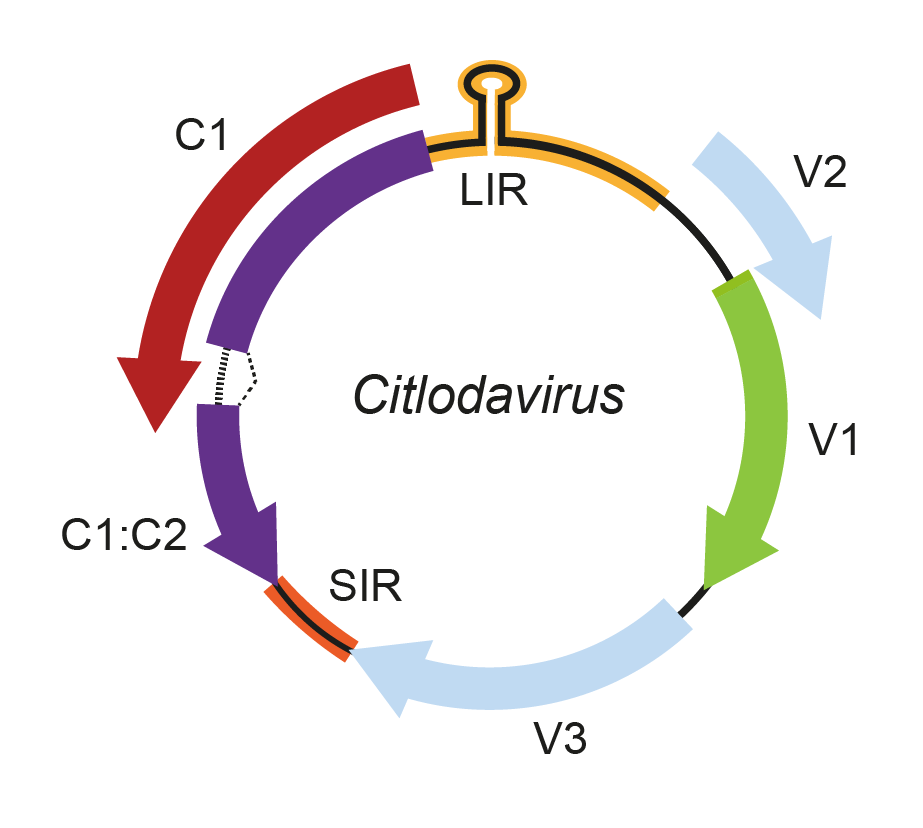
Like mastreviruses, capulaviruses and grabloviruses, the genome of citlodaviruses contains two intergenic regions, the long intergenic region (LIR) where the v-ori and transcription start sites are located, and the short intergenic region (SIR) that would contain the complementary-strand replication origin and transcription termination sites.
Biology
Host range
Members of species from this genus have a limited natural host range. They have been isolated from symptomatic dicot trees and shrubs. Camellia chlorotic dwarf-associated virus has been isolated from common camellia (Camellia japonica) (Zhang et al., 2018), citrus chlorotic dwarf associated virus from citrus trees (Citrus spp.) (Loconsole et al., 2012), paper mulberry leaf curl virus 2 (Qiu et al., 2020) from paper mulberry (Broussonetia papyrifera) and passion fruit chlorotic mottle virus from passion fruit (Passiflora edulis) (Fontenele et al., 2018). An infectious clone of passion fruit chlorotic mottle virus systemically infected Nicotiana benthamina and Arabidopsis thaliana in addition to passion fruit (Fontenele et al., 2018).
Species demarcation criteria
Isolates of species in the genus share less than 63% nucleotide sequence identity with all other known geminiviruses. The tentative species demarcation criteria of 78% have been defined, but this value could change when additional citlodavirus sequences become available.