

Family: Arenaviridae
Genus: Hartmanivirus
Distinguishing features
Like reptarenaviruses, hartmaniviruses infect captive snakes. In contrast to mammarenaviruses and reptarenaviruses but similar to antennaviruses and innmoviruses, hartmaniviruses lack a gene encoding the zinc-binding protein (Z) (Hepojoki et al., 2018).
Virion
Morphology
Virions are spherical or pleomorphic in shape, 120–150 nm in diameter, with dense lipid envelopes (Figure 1.Hartmanivirus). The virion surface layer is covered with club-shaped projections. These projections are made of trimeric spike structures of two virus-encoded membrane glycoprotein (GP) subunits (GP1 and GP2) and a stable signal peptide (SSP) (Hepojoki et al., 2018).
 |
| Figure 1.Hartmanivirus. Hartmanivirus particles. (A) Cryo-electron micrograph of Haartman Institute snake virus 1 (HISV1). Courtesy of Pasi Laurinmäki and Sarah Butcher, Cryo-EM Core Facility, Biocenter Finland, University of Helsinki, Finland. (B) Negative-stain electron micrograph of HISV1. Courtesy of Andrea Laimbacher and Elisabeth M. Schraner, Institute of Veterinary Anatomy and Virology, Vetsuisse Faculty, University of Zurich, Switzerland. |
Physicochemical and physical properties
Not reported
Nucleic acid
Hartmaniviruses have an ambisense RNA segment and a negative-sense RNA segment that are encapsidated independently. The termini of the RNAs contain inverted complementary sequences encoding transcription and replication initiation signals (Hepojoki et al., 2018).
Proteins
Hartmaniviruses express three structural proteins. The most abundant structural protein in hartmanivirions is nucleoprotein (NP), which encapsidates the genomic segments. The least abundant protein is the large (L) protein, which mediates virus genome replication and transcription. Glycoprotein subunits GP1 and GP2 are derived by post-translational cleavage from an intracellular GP precursor (GPC). A third GPC cleavage product, the SSP, stays attached to the GP complex (Hepojoki et al., 2018).
Lipids
Not reported (SSP is likely myristoylated (Hepojoki et al., 2018).)
Carbohydrates
Not reported
Genome organization and replication
The S and L RNAs of hartmaniviruses encode proteins in non-overlapping open reading frames (ORFs) of opposite polarities (ambisense coding arrangement) that are separated by non-coding intergenic regions (IGRs) (Figure 2.Hartmanivirus). The S RNA encodes NP in the virus genome-complementary sequence and the GPC in the virus genome-sense sequence. The L RNA encodes the L protein in the virus genome-complementary sequence. The IGRs form one or more energetically stable stem-loop (hairpin) structures and function in structure-dependent transcription termination and in virion assembly and budding (Hepojoki et al., 2018).
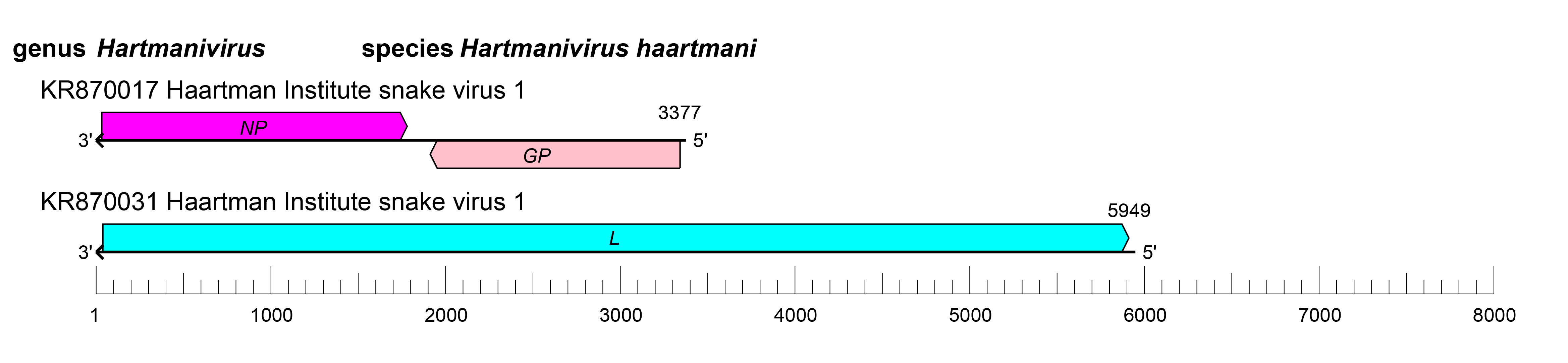 |
| Figure 2.Hartmanivirus. Schematic representation of hartmanivirus genome organization. The 5′- and 3′-ends of both segments (small [S] and large [L]) are complementary at their termini, likely promoting the formation of circular ribonucleoprotein (RNP) complexes within the virion. GP, glycoprotein gene; L, large protein gene; NP, nucleoprotein gene. |
Biology
Hartmaniviruses were discovered in Helsinki, Finland, in a captive boa constrictor (Squamata: Boidae: Boa constrictor Linnaeus, 1758) that was co-infected with a reptarenavirus and had boid inclusion body disease (BIBD) (Hepojoki et al., 2015). Several distinct hartmaniviruses have been isolated in boa constrictor I/1Ki cells. Whether hartmaniviruses cause disease in snakes remains to be determined (Hepojoki et al., 2018).
Species demarcation criteria
The parameters used to assign viruses to new species in the genus are:
- the virus shares less than 80% nucleotide sequence identity in the S RNA segment and less than 76% identity in the L RNA segment with other viruses;
- association of the virus with a distinct main host or a group of sympatric hosts;
- dispersion of the virus in a distinct defined geographical area; and/or
- the virus shares less than 88% NP amino-acid sequence identity with other viruses (Radoshitzky et al., 2015).
Relationships within the genus
Phylogenetic relationships across the genus have been established from maximum-likelihood trees generated using complete sequences of NP and L proteins (Figure 3.Hartmanivirus).
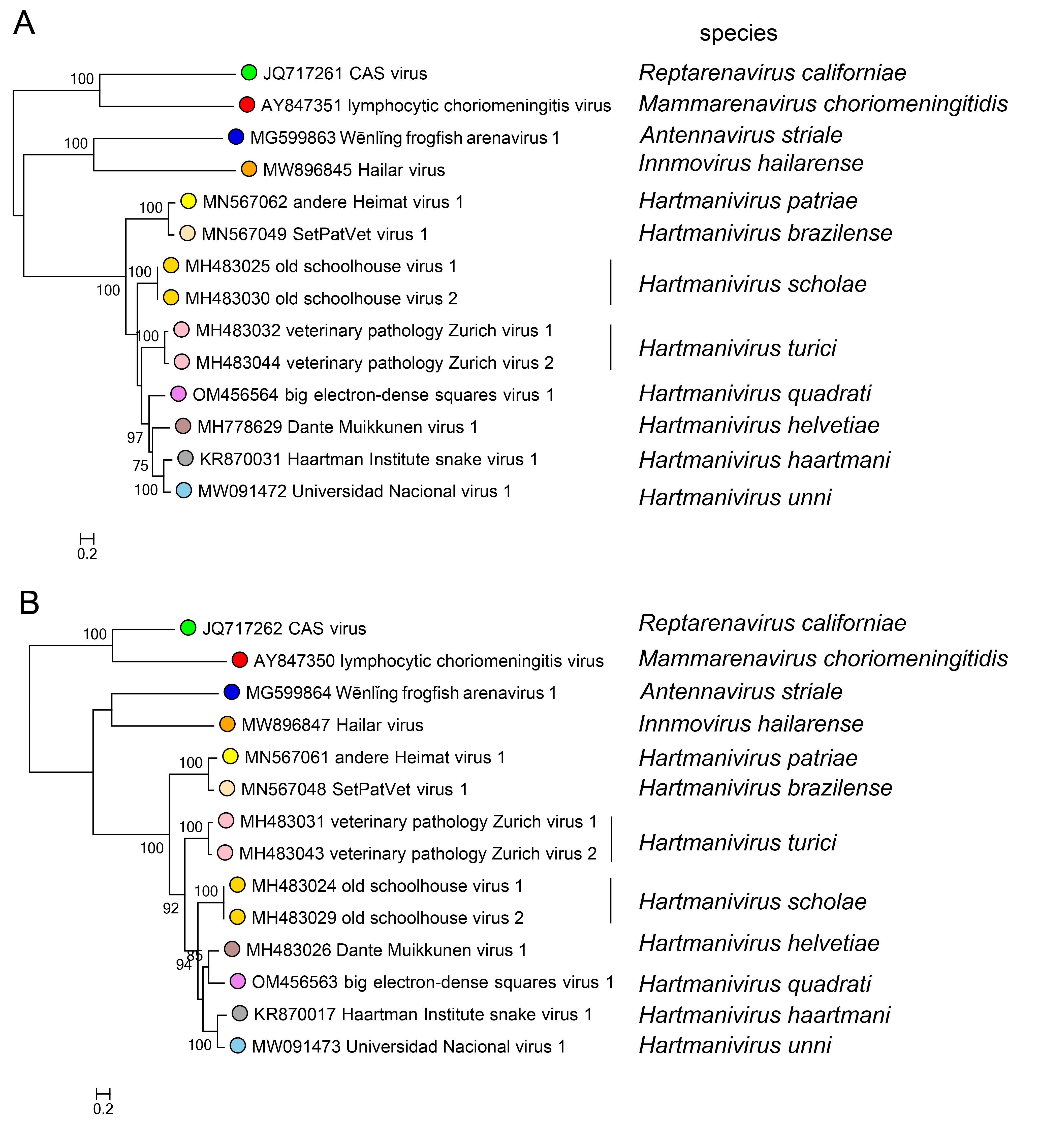 |
| Figure 3.Hartmanivirus. Maximum-likelihood phylogenetic trees inferred from MAFFT alignments (Katoh and Standley 2013) of the large (L) (A) and nucleoprotein (NP) (B) amino-acid sequences. The trees were generated by the IQ-TREE software v.1.6.12 (Trifinopoulos et al., 2016) using the best-fitting evolutionary model. Branch supports were calculated using the ultrafast bootstrap method (1,000 bootstraps). The mid-point rooted trees were visualized using FigTree (http://tree.bio.ed.ac.uk/). For L protein and NP, 10 classified viruses (dark green dots) were included. In both trees, representative viruses of the genera Mammarenavirus (red dot), Reptarenavirus (light green dot), Antennavirus (cyan dot), and Innmovirus (yellow dot) are also included. |