

Family: Coronaviridae
Patrick C.Y. Woo, Raoul J. de Groot, Bart Haagmans, Susanna K.P. Lau, Benjamin W. Neuman, Stanley Perlman, Isabel Sola, Lia van der Hoek, Antonio C.P. Wong and Shiou-Hwei Yeh
The citation for this ICTV Report chapter is the summary published as:
Corresponding author: Patrick C.Y. Woo (E-mail: [email protected])
Edited by: Peter Simmonds and Stuart G, Siddell
Posted: February 2023
Summary
Members of the family Coronaviridae, a monophyletic group of viruses in the order Nidovirales, are enveloped, positive-sense RNA viruses that are known to infect four of the seven classes of vertebrates: mammals and birds (orthocoronaviruses), amphibians (letoviruses) and bony fish (pironaviruses); (Table 1 Coronaviridae). In terms of genome size and genetic complexity, members of the family Coronaviridae, are among the largest RNA viruses identified so far. RNA viruses with larger genomes are also members of the Nidovirales, including members of the species Alphaabyssovirus aplyiaes (35.9 kb, family Abyssoviridae) and Alphamononivirus schmidteae (41.1 kb, family Mononiviridae), which infect invertebrates. Replication has been studied in detail only for orthocoronaviruses. Orthocoronavirus virions attach to host cell surface receptors via their spikes and release their genome into the target cell cytoplasm via fusion of the viral envelope with the plasma membrane or the limiting membrane of endocytic vesicles. Members of the family Coronaviridae infect humans and a variety of animals resulting in diverse clinical manifestations, ranging from asymptomatic to severe fatal diseases. Members of two species of orthocoronavirus, Betacoronavirus pandemicum and Betacoronavirus cameli, are highly pathogenic to humans, leading to the SARS and MERS epidemics, and the COVID-19 pandemic.
Table 1 Coronaviridae Characteristics of members of the family Coronaviridae
| Characteristic | Description |
| Example | murine hepatitis virus A59 (AY700211), species Betacoronavirus muris |
| Virion | Enveloped, pleomorphic but often quasi-spherical, 80–160 nm with apparent spike projections |
| Genome | 22–36 kb of positive-sense RNA |
| Replication | Through an antigenomic RNA generated by continuous transcription; gene expression through discontinuous transcription of a nested set of co-terminal subgenomic negative-sense RNAs which are copied into subgenomic mRNAs |
| Translation | From capped and polyadenylated genomic and subgenomic mRNAs |
| Host range | Vertebrates (mammals, birds, amphibians and fish) |
| Taxonomy | Realm Riboviria, kingdom Orthornavirae, phylum Pisuviricota, class Pisoniviricetes, order Nidovirales, suborder Cornidovirineae, family Coronaviridae; the family includes 3 subfamilies (Letovirinae, Orthocoronavirinae and Pitovirinae), 6 genera, 28 subgenera, and 54 species |
Virion
Morphology
Virions of members of the subfamily Orthocoronavirinae are spherical, 80–160 nm, enveloped particles decorated with large club- or petal-shaped surface projections (the “peplomers” or “spikes”), which in electron micrographs of spherical particles create an image reminiscent of the solar corona (Barcena et al., 2009) (Figure 1 Coronaviridae). This inspired the name “coronaviruses”, originally for the viruses now grouped in the subfamily Orthocoronavirinae and later adopted for the whole family. Orthocoronavirus virion assembly is achieved by budding of preformed nucleocapsids at smooth intracellular membranes of endoplasmic reticulum/early Golgi compartments (Perlman and Netland 2009). Members of the subgenus Embecovirus (genus Betacoronavirus) display a second type of surface projection, 5–7 nm in length, comprised of the homodimeric hemagglutinin-esterase (HE) glycoprotein (Figure 1E.Coronaviridae) (Hurdiss et al., 2020).
Orthocoronavirus virions studied by cryo-electron tomography are homogeneous in size and spherical (envelope outer diameter 85±5 nm) (Barcena et al., 2009). The envelope is exceptionally thick (7.8±0.7 nm) in comparison to typical biological membranes (average thickness 4 nm) (Barcena et al., 2009), and is packed with a matrix network consisting of membrane protein dimers (Neuman et al., 2011). The viral ribonucleoprotein consists of the RNA genome and nucleoprotein (N), and is organized in short helical units that are arranged loosely in the virion interior (Yao et al., 2020), but can be appended to the inner surface of the envelope at a distance of about 4 nm (Figure 1 Coronaviridae) (Chang et al., 2006).
For members of the subfamily Letovirinae, the shape and size of virions are unknown, but downstream open reading frames (ORFs) encode a large spike (S)-like protein, a three-pass transmembrane protein that resembles the coronavirus membrane (M) protein, an envelope (E)-like protein and a putative nucleocapsid (N) protein (Bukhari et al., 2018). Several of these putative structural proteins have predicted transmembrane regions, suggesting that virions are enveloped (Bukhari et al., 2018).
For members of the subfamily Pitovirinae, the shape and size of virions are similarly undetermined. However, the genome of members of the species Alphapironavirus salmonis (for example Pacific salmon nidovirus, MK611985) encodes proteins putatively identified as similar to the coronavirus S, M, E and N proteins that may create virions morphologically related to those of other coronaviruses.
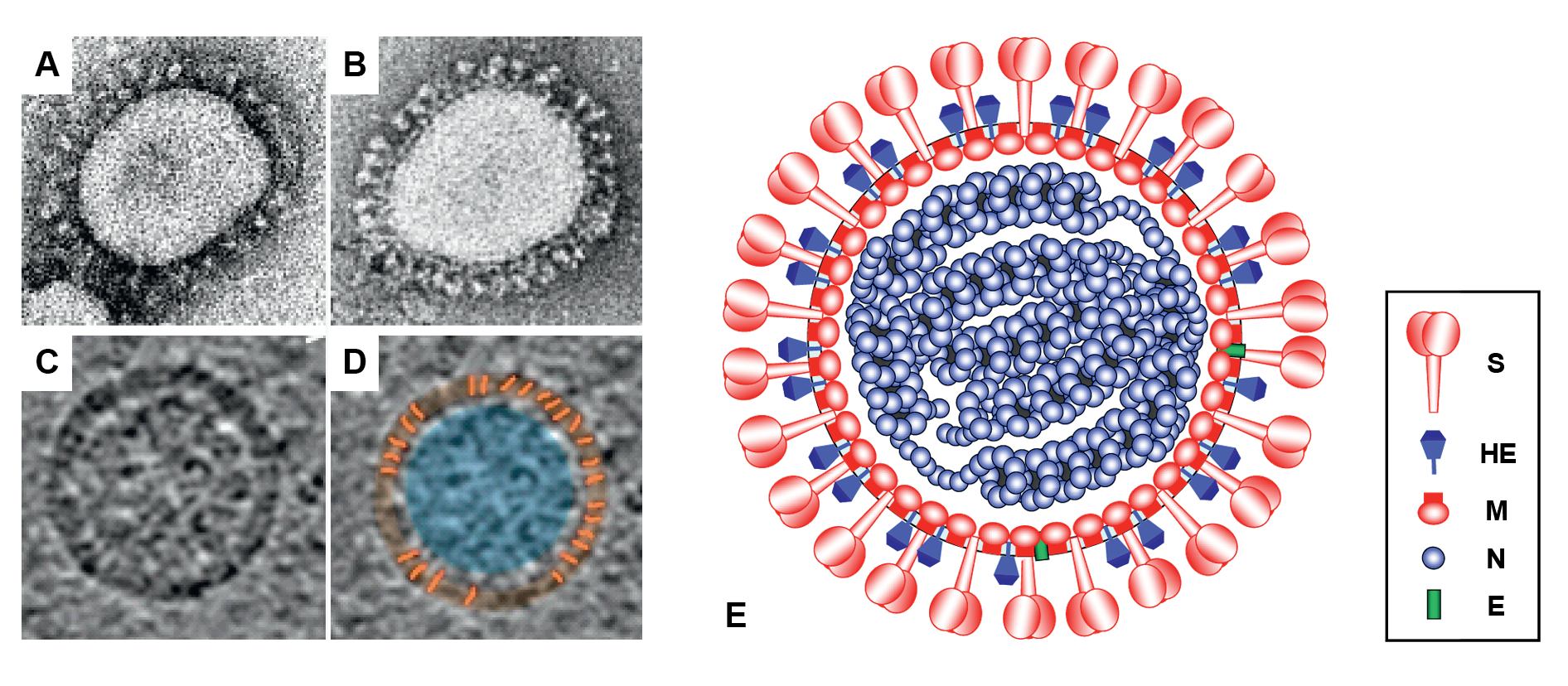 |
| Figure 1.Coronaviridae Coronavirus virion morphology. (A, B) Negative-staining (2% phosphotungstic acid) electron micrographs of (A) a virion of murine hepatitis virus (MHV) laboratory strain A59 that lacks HE expression and (B) a recombinant MHV-A59 virus in which HE expression was restored (courtesy Jean Lepault, Laboratory of Molecular and Structural Virology, Gif-sur-Yvette Cedex, France). (C, D) Cryo-electron tomographs of MHV. A virtual slice (7.5 nm thick) through a reconstructed MHV particle (left) with highlighted features superimposed (right). The envelope is colored in orange with conspicuous striations highlighted; the nucleocapsid region is colored in blue. Note low-density region (ca. 4 nm) between envelope and nucleocapsid (reprinted with permission from (Barcena et al., 2009) (E) Schematic representation of an embecovirus virion. |
Physicochemical and physical properties
For members of the subfamily Orthocoronavirinae, the estimated Mr of the virion is 400×106, its buoyant density in sucrose and CsCl is 1.15–1.20 g cm−3 and 1.23–1.24 g cm−3, respectively, and its S20,W is 300–500S (Gorbalenya et al., 2006). Particles are sensitive to heat, lipid solvents, non-ionic detergents, formaldehyde, oxidizing agents and UV irradiation (Bedell et al., 2016, Patterson et al., 2020).
No information is available for members of the subfamilies Letovirinae and Pitovirinae.
Nucleic acid
Genomes of members of the family Coronaviridae consist of positive-sense, linear and infectious RNA of 22–36 kb that is capped, polyadenylated and polycistronic (Barnard 2008). For the subfamily Orthocoronavirinae, the RNA is 26–31 kb and is infectious (Fehr and Perlman 2015).
The only letovirus genome studied is an assembly of 22 304 nucleotides, potentially encoding proteins equivalent to those of severe acute respiratory syndrome coronavirus (SARS-CoV) from the nsp3 gene to the 3′-end (Bukhari et al., 2018). This sequence is missing the 5′-terminal sequences of orthocoronaviruses, including a 5′-non-coding region and sequences corresponding to coronavirus nsp1 to part of nsp3 (Bukhari et al., 2018). The missing genomic region is 1500–4000 nucleotides compared to the complete genomes of the small deltacoronaviruses or the relatively large alphacoronaviruses (Bukhari et al., 2018). The letovirus genome contains a 572-nucleotide 3′-non-coding region and an 18-nucleotide poly-adenosine tail. The start codons of the putative S and M ORFs overlap with the stop codons of preceding ORFs, suggesting a relatively compact genome (Bukhari et al., 2018).
The only available pirovirus sequence (Pacific salmon nidovirus, MK611985) studied is 36 652 nucleotides with an internal sequence gap located after the end of ORF1b and before the beginning of the 3′-terminal region encoding the putative structural and accessory proteins, which raises the possibility of this being a bipartite genome. The large genome is partly due to the presence of a large putative accessory gene of unknown function. The available genome contains a 22-nucleotide poly-adenosine tail.
Proteins
Orthocoronaviruses all share the following structural proteins:
- the spike protein (S), a large homo-trimeric type I membrane glycoprotein. S is a class I fusion protein that mediates receptor-binding and membrane fusion (Bosch et al., 2003, Alsaadi et al., 2019).
- the membrane glycoprotein (M), an integral type III membrane protein with predicted triple-spanning NexoCendo topology (Locker et al., 1992). Depending on the virus species, the amino-terminal ectodomain is decorated with N- or O-linked glycans (Voss et al., 2009). The long C-terminal endodomain, comprising an amphiphilic region and a hydrophilic tail, is believed to associate with the inner leaflet of the membrane to form a matrix-like lattice, which would explain the remarkable thickness of the orthocoronavirus envelope (Figure 1 Coronaviridae) (de Haan et al., 1998). In transmissible gastroenteritis virus (TGEV, species Alphacoronavirus suis) infected pigs, a second population of tetra-spanning M proteins, adopting an Nexo-Cexo topology in the viral envelope, has been described (Escors et al., 2001); M can adopt two conformations, which are elongated and compact M proteins (Neuman et al., 2011). The elongated form of M protein is responsible for the formation of a convex, rigidified viral envelope (Neuman et al., 2011).
- the envelope protein (E), a small pentameric integral membrane protein with ion channel and/or membrane permeabilizing (viroporin) activities (Ye and Hogue 2007). With around 20 copies per particle, the E protein is only a minor structural component (Godet et al., 1992). Although its precise function remains to be defined, the E plays a role in virion assembly and morphogenesis and has been identified as a virulence factor for some viruses including severe acute respiratory syndrome coronavirus (SARS-CoV) (Perlman and Netland 2009, DeDiego et al., 2011, Fett et al., 2013, Alsaadi et al., 2020).
- the nucleocapsid protein (N), an RNA-binding phosphoprotein. ADP-ribosylation of N protein is observed during infection (Grunewald et al., 2018). Besides its obvious function in genome encapsidation, the N protein is also involved in RNA synthesis and translation, displays RNA chaperone activity, and acts as a type I interferon antagonist (Enjuanes et al., 2006, Saikatendu et al., 2007, Wu et al., 2009, Zuniga et al., 2010). Nucleocapsids are helical and can be released from the virion by treatment with detergents (Chen et al., 2007). The orthocoronavirus nucleocapsid appears to be loosely-wound, with small helical units distributed throughout the virion interior (Yao et al., 2020), that of letovirus remains to be characterised (Bukhari et al., 2018).
Members of different orthocoronavirus species can have additional accessory proteins incorporated into the virion. For example, members of the subgenus Embecovirus in the genus Betacoronavirus code for an accessory homo-dimeric type I envelope glycoprotein, the hemagglutinin-esterase (HE). This protein mediates reversible virion attachment to O-acetylated sialic acids by acting both as a lectin and as a sialate-O-acetylesterase (Zeng et al., 2008, Langereis et al., 2010). The HE of MHV shares about 30% aa sequence identity with the HE protein of toroviruses (family Tobaniviridae, which are also classified in the order Nidovirales to which coronaviruses are assigned). However, it is equally related to subunit 1 of the hemagglutinin-esterase fusion protein (HEF) of the structurally dissimilar influenza C virus (family Orthomyxoviridae) (Zeng et al., 2008). In virions of MHV, the stoichiometric ratio of N, M and HE proteins is approximately 1 : 2.6 : 0.4; in TGEV, N and M occur at a ratio of 1 : 3. There are no reliable estimates for the S protein as it is present in small quantities in virus particles, may occur both in cleaved and uncleaved forms, and is easily lost during virus purification.
Members of the family Coronaviridae all seem to share three envelope protein types, the envelope (E), membrane (M) and spike (S) proteins, which are essential for virion morphogenesis or infectivity or both (Barcena et al., 2009, Yuan et al., 2017). Similarities in size, predicted structures and presumed functions suggest a common ancestry, and the distant but clear sequence similarities observed between letovirus and orthocoronavirus S proteins lend further support to this view (Bukhari et al., 2018).
The 3′-proximal region of the letovirus genome contains six ORFs that could encode proteins of 50 or more amino acids, including the viral structural proteins (Bukhari et al., 2018). The first ORF encodes a large S-like protein of 1526 amino acids with an amino-terminal signal peptide and a carboxyl-terminal transmembrane region (Bukhari et al., 2018). The second and third ORFs encode respectively, a unique single-pass transmembrane protein of 55 amino acids and a unique soluble 157-amino acid protein; these are likely strain-specific accessory proteins (Bukhari et al., 2018). The fourth ORF encodes an E-like protein of 77 amino acids with an amino-terminal predicted transmembrane region followed by a potential amphipathic helix (Bukhari et al., 2018). The fifth ORF encodes a 241-amino acid three-pass transmembrane protein resembling coronavirus M protein. The sixth ORF putatively encodes an N protein of 459 amino acids (Bukhari et al., 2018).
The 3′-proximal region of the pironavirus genome contains five ORFs that are predicted to encode proteins of 130 or more amino acids, including the viral structural proteins. The first ORF encodes a large lamina-associated polypeptide 1C (LAP1C)-like protein of 2038 amino acids. The second ORF encodes a S-like protein of 1213 amino acids. The third ORF encodes an E-like protein of 132 amino acids. The fourth ORF encodes a 446-amino acid M-like protein. The fifth ORF putatively encodes an N protein of 513 amino acids
Lipids
Orthocoronaviruses acquire their lipid envelopes by budding at membranes of the endoplasmic reticulum, intermediate compartment and/or Golgi complex of the host cells (Klumperman et al., 1994). The S and E proteins are palmitoylated (Corse and Machamer 2002, Liao et al., 2006, Petit et al., 2007, Lopez et al., 2008).
No information is available for members of the subfamilies Letovirinae and Pitovirinae.
Carbohydrates
The S protein of orthocoronaviruses and the HE protein of members of the subgenus Embecovirus are heavily glycosylated and contain multiple N-linked glycans (20–35 and 5–11, respectively) (de Groot 2006, Zeng et al., 2008). The M protein of orthocoronaviruses contains a small number of either N- or O-linked glycans, depending on the virus, located near the amino-terminus (Holmes et al., 1981, Voss et al., 2006). The nsp3 ectodomains of SARS-CoV and MHV are N-linked glycosylated (Harcourt et al., 2004, Kanjanahaluethai et al., 2007). The ORF3 accessory protein of human coronavirus NL63 (HCoV-NL63) is N-glycosylated at the N-terminus (Muller et al., 2010). Orthocoronavirus E proteins are not glycosylated.
No information is available for members of the subfamilies Letovirinae and Pitovirinae.
Genome organization and replication
Members of the family Coronaviridae have genomes with multiple ORFs (Figure 2 Coronaviridae) in the order 5′-NCR-replicase-S-E-M-N-NCR-3′ (genes are named after their product) (Figure 3.Coronaviridae), with the genome functioning as mRNA for the replicase gene (de Vries et al., 1997). The replicase gene is comprised of two overlapping ORFs called 1a and 1b that occupy the 5′-two thirds of the genome. The replicase gene is translated to produce the polyprotein pp1a and, following programmed −1 ribosomal frameshifting, a C-terminally extended product, pp1ab (Bredenbeek et al., 1990). The polyproteins are co- and post-translationally processed by virus-encoded proteinases and, thus, are not detectable as full-length proteins in virus-infected cells (Graham and Denison 2006). The N-termini of pp1a and pp1ab are processed by one or two papain-like proteinases, whereas the C-terminal half of pp1a and the ORF1b-encoded part of pp1ab are cleaved at 11 well-conserved sites by the main proteinase (Mpro or 3CLpro), an enzyme conserved within members of the order Nidovirales with a chymotrypsin-like fold that has a poliovirus 3C proteinase-like substrate specificity with a cysteine as the active site nucleophile (Ziebuhr et al., 2000, Ziebuhr 2005). Proteolytic processing of the replicase protein results in the production of 14 to 16 mature products, commonly referred to as non-structural proteins (nsps) and numbered according to their position from the N-terminus of the virus polyprotein (Figure 2 Coronaviridae). Many nsps are unique enzymes involved in one or more essential steps in viral replication. Others appear to be exclusively involved in virus–host interactions (including immune evasion) and are dispensable for virus propagation in vitro (Table 2 Coronaviridae).
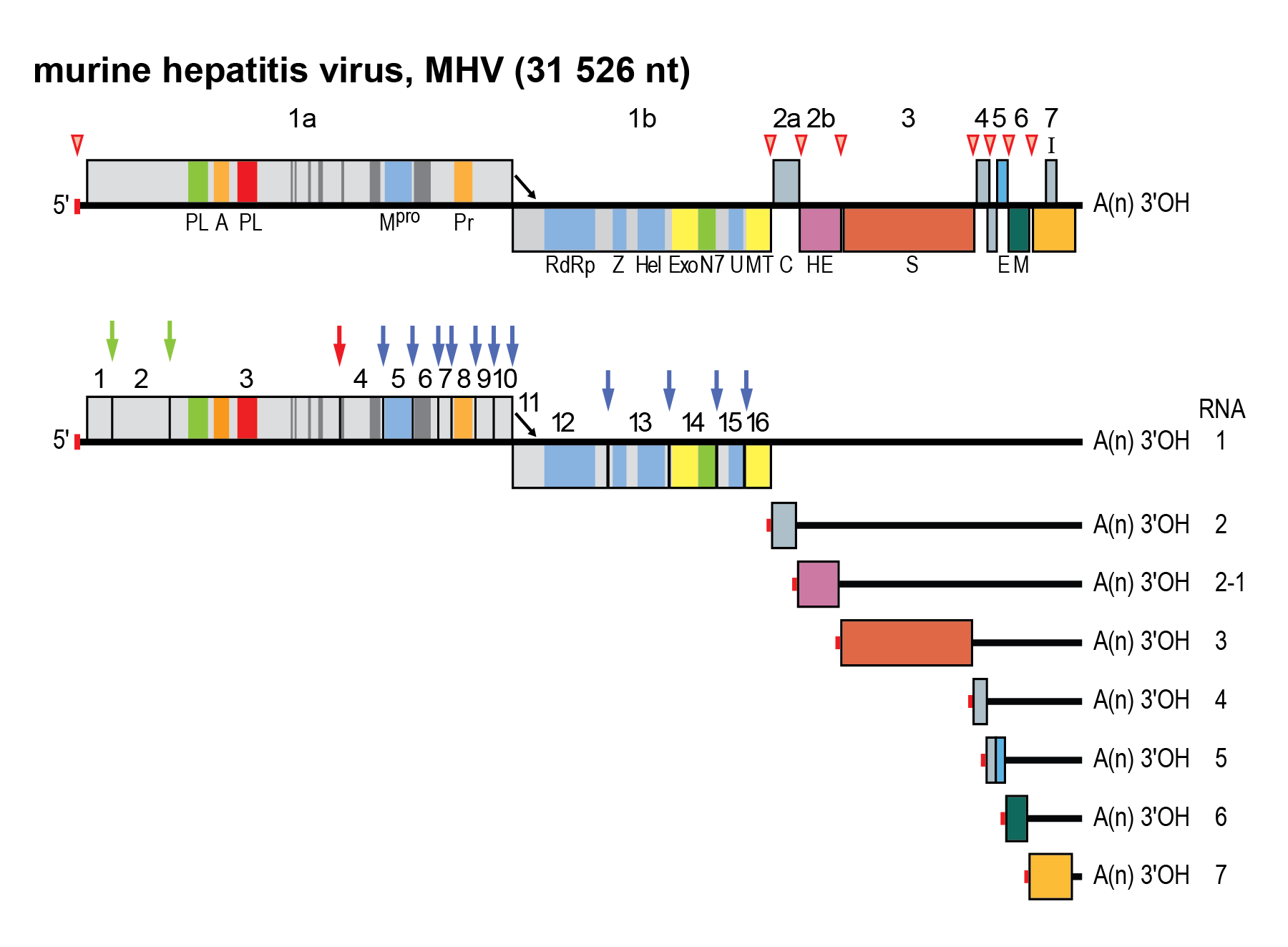 |
| Figure 2.Coronaviridae. Coronavirus genome organization and expression. (Upper panel). Schematic representation of the genome of MHV. ORFs are represented by boxes, indicated by number (above) and encoded protein (acronyms below). Regions encoding key domains in replicase polyproteins pp1a and pp1ab are colour-coded with hydrophobic segments shown in dark grey. The 5′-leader sequence is depicted by a small red box. The arrow between ORF 1a and 1b represents the ribosomal frameshifting site. The poly(A) tail is indicated by “A(n)”. Red arrowheads indicate the locations of transcription-regulating sequences (TRSs). PL (green) papain-like proteinase 1 (PL1pro); PL (red), papain-like proteinase 2 (PL2pro); A, ADP-ribose-1″phosphatase (macrodomain); Mpro, 3C-like main protease; Pr, noncanonical RNA-dependent RNA polymerase, putative primase; RdRP, RNA-dependent RNA polymerase; Z, zinc-binding domain; Hel, helicase domain; Exo, 3′-to-5′ exoribonuclease domain; N7, guanine-N7-methyltransferase; U, nidoviral uridylate-specific endoribonuclease (NendoU); MT, ribose-2′-O-methyltransferase domain; HE, hemagglutinin-esterase; S, spike protein; E, envelope protein; M, membrane protein, N, nucleocapsid protein. (Lower panel) Processing of the replicase polyproteins and structural relationship between the genomic RNA and subgenomic mRNAs of coronaviruses. Arrows indicate cleavage sites for PL1pro (green), PL2pro (red) and Mpro (blue). The locations of the non-structural proteins (nsp’s) are indicated by their number (Table 2 Coronaviridae). mRNA species are numbered as by convention on the basis of their size, from large to small, with the genome designated as RNA1. For the sg mRNAs only ORF(s) that are translated are shown. |
 |
| Figure 3 Coronaviridae Genome organizations of selected viruses from each genus. Whole genome organizations with ORFs are depicted as coloured boxes with rosybrown, ORF1a; pink, ORF1b; red, S; cyan, E; yellow, M and magenta, N and skyblue, lamina-associated polypeptide 1C (LAP1C). ORFs for accessory proteins are depicted as coloured boxes in grey. |
Table 2 Coronaviridae Cleavage products of orthocoronavirus replicase polyproteins pp1a and pp1ab: names and assigned functions*
| Protein | Assigned function |
|---|---|
| nsp1† | IFN antagonist |
| Degradation of host mRNAs | |
| Inhibition of translation | |
| Cell cycle arrest | |
| nsp2 | Unknown; associates with RTCs |
| nsp3 | Papain-like proteinase PL1pro; polyprotein processing |
| Papain-like proteinase PL2pro; polyprotein processing, DUB | |
| ADP-ribose-1″phosphatase (macrodomain); RNA-binding; N protein binding | |
| IFN antagonist | |
| DMV formation | |
| nsp4 | DMV formation |
| nsp5 | 3C-like protease (3CLpro); polyprotein processing |
| nsp6 | DMV formation |
| nsp7 | ssRNA binding; Subunit of RdRP holoenzyme |
| nsp8 | Subunit of RdRP holoenzyme; Putative primase; Forms hexadecameric supercomplex with nsp7; Putative 3′-terminal adenylyltransferase |
| nsp9 | Putative ssRNA binding; associates with RTCs; involved in RNA capping |
| nsp10 | Dodecameric zinc finger protein; associates with RTCs, stimulates nsp14 exonuclease and nsp16 methyltransferase activity; regulation of ribosomal frameshifting |
| nsp11 | Unknown |
| nsp12 | RdRP; NiRAN domain (NMPylase and putative RNA-capping enzyme) |
| nsp13 | Zinc-binding domain-containing Helicase |
| RNA 5′-triphosphatase | |
| nsp14 | 3′→5′exoribonuclease (required for RdRP fidelity) |
| Guanine-N7-methyltransferase (RNA cap formation) | |
| nsp15 | Hexameric uridylate-specific endoribonuclease |
| nsp16 | Ribose-2′-O-methyltransferase (RNA cap formation) |
IFN, interferon; RTC, replicase/transcriptase complex; DUB, deubiquitinating enzyme; DMV, double-membrane vesicles; NMP, nucleoside monophosphate.
* references: (Joseph et al., 2006, Su et al., 2006, Joseph et al., 2007, Neuman et al., 2008, Perlman and Netland 2009, Angelini et al., 2013, Becares et al., 2016, Fehr et al., 2016, Doyle et al., 2018, V'Kovski et al., 2021, Park et al., 2022b)
† Absent in gammacoronaviruses and deltacoronaviruses.
The replicase polyproteins of members of the family Coronaviridae comprise a number of characteristic domains arranged in a conserved order (Figure 2 Coronaviridae and Table 2 Coronaviridae). Two ORF1-encoded replicase domains, an ADP-ribose-1″-phosphatase (ADRP, also called macrodomain; located in orthocoronavirus nsp3 but partially truncated in the letovirus genome) and a noncanonical “secondary” RNA-dependent RNA polymerase (RdRP) with possible primase activity (nsp8) may represent diagnostic markers that distinguish members of the family Coronaviridae from members of other families in the order Nidovirales (Imbert et al., 2006, Bukhari et al., 2018).
The entire viral replication cycle takes place in the cytoplasm and involves two processes, the production of full-length genomic RNAs (gRNA), known as replication, and the synthesis of subgenome-sized mRNAs (sgmRNA), known as transcription. The replication and transcription processes involve the generation of genome-length negative-sense strand and subgenomic negative-sense RNA intermediates, respectively (Sawicki and Sawicki 2005). Genomic and subgenomic negative-sense RNA molecules are produced from full length genomic RNA and serve as the templates for the synthesis of genomic and subgenomic mRNAs, respectively (Sawicki and Sawicki 2005).
The structural basis for orthocoronavirus RNA synthesis is increasingly well understood, though questions remain about the order of some steps in the process. RNA synthesis is catalyzed by a replication–transcription complex (RTC) that contains proteins from pp1a and pp1b, in complexes that are rearranged to carry out RNA synthesis and capping activities (Yan et al., 2021). During synthesis of positive-sense strand viral RNAs, the NiRAN domain of the viral RNA-dependent RNA polymerase (nsp12 or RdRP) transfers one or more nucleotides to nsp9, which is then involved in transfer of the cap structure to the viral RNA (Lehmann et al., 2015, Slanina et al., 2021, Park et al., 2022a). Viral RNA synthesis then involves dual polymerase activities of the nsp7+nsp8 complex and the main viral polymerase, nsp12 (te Velthuis et al., 2012). RNA cap methylation is carried out by complexes of nsp10, nsp14 and nsp16 (Chen et al., 2009, Chen et al., 2011). The orthocoronavirus replication-transcription complex is composed of viral and host proteins and associated with an interconnected network of modified intracellular membranes and double-membrane vesicles that are presumably endoplasmic reticulum (ER)-derived (Gosert et al., 2002, Al-Mulla et al., 2014, Sola et al., 2015), with access to the interior of the double-membrane vesicles via a molecular pore that contains at least some of the transmembrane domain-containing cleavage products from pp1a: nsp3, nsp4 and nsp6 (Wolff et al., 2020). Understanding of the replication cycle of letoviruses and piroviruses is limited. The existence of subgenomic mRNAs of M and N genes of letoviruses suggests these two genes are expressed (Bukhari et al., 2018). Similar genomic organization between orthocoronaviruses and letoviruses, including conservation of nsp3–16, suggest that they may use similar replication strategies and intracellular modifications (Bukhari et al., 2018).
Replication of the genomic RNA uses a full-length negative-sense RNA as an intermediate. The 3′-proximal genes (putatively 6 in letoviruses and up to 12 in some orthocoronaviruses) code for the structural proteins and, in the case of orthocoronaviruses, a variable number of “accessory” or “niche-specific” proteins. These genes are expressed – as is typical for nidoviruses – from a 3′-coterminal nested set of capped and polyadenylated subgenomic mRNAs (Sethna et al., 1989, Sawicki and Sawicki 1990, Sethna et al., 1991).
For orthocoronaviruses, virus assembly involves budding of preformed nucleocapsids at membranes of the endoplasmic reticulum and early Golgi compartment and the completed virions are released via the exocytotic pathway (de Haan and Rottier 2005, Stertz et al., 2007). A recent study suggests that betacoronaviruses can also utilize lysosome-mediated cell egress pathway (Ghosh et al., 2020). The viral assembly mechanism remains to be elucidated for members of the subfamilies Letovirinae and Pitovirinae.
Orthocoronavirus genomes contain 5′- and 3′- NCRs of 200–600 nt and 200–500 nt, respectively. Signals for genome replication and encapsidation reside not only in these NCRs, but also in adjacent and more internal coding regions (Brian and Baric 2005). Six ORFs are conserved family-wide and arranged in a fixed order: (as listed in the 5′-to-3′ direction) ORFs 1a and 1b, together comprising the replicase genes, and the ORFs for the structural proteins S, E, M and N (de Vries et al., 1997). Downstream of ORF1b and interspersed between the structural protein genes, there exist various numbers of accessory genes, the products of which are generally dispensable for replication in vitro, but affect replication or pathogenicity during natural infection (Figure 2 Coronaviridae) (Zhao et al., 2009, Niemeyer et al., 2013, Fehr and Perlman 2015, Rabouw et al., 2016, Canton et al., 2018, Castano-Rodriguez et al., 2018, Schroeder et al., 2021).
These accessory genes may have been acquired through horizontal gene transfer and occasionally lost as viruses have adapted to new hosts and niches (Wu et al., 2016b). The diversity of accessory genes between orthocoronavirus subgenera, species or strains attest to the plasticity and highly dynamic nature of the 3′-proximal third of the orthocoronavirus genome.
While the genome serves as mRNA for the replicase polyproteins, the 3′-proximal genes are expressed from a nested set of subgenomic mRNAs, the coding regions of which (the “body” sequences) are 3′-coterminal with the genome (Tijms et al., 2001). Each of these mRNAs has a short 5′-leader sequence identical to the 5′-terminal end of the genome. All except the smallest mRNAs are structurally polycistronic but translation is restricted to the 5′-proximal ORF(s) (Figure 4 Coronaviridae).
Coronavirus discontinuous transcription is driven by short conserved sequence (CS) elements, included in a larger and more variable region commonly called the transcription-regulatory sequence (TRS) (Dufour et al., 2011, Fehr and Perlman 2015) and depends on multiple factors including RNA-RNA and RNA-protein interactions (Sola et al., 2015). A TRS copy is found at the 5′-end of the genome, immediately downstream of the leader sequence and preceding 3′-proximal ORFs. According to the prevailing model for transcription, leader–body fusion occurs during the 3′-discontinuous extension of subgenomic negative-sense RNAs through a template switch of the replication-transcription complex from 3′-end TRSs to the leader TRS on the genome template (Zuniga et al., 2004, Sola et al., 2005). Transcription template-switch resembles homology-assisted RNA recombination (Smits et al., 2005). This process is driven by sequence complementarity between the anti-TRS at the 3′-end of the nascent negative-sense RNA and the 5′-leader TRS (Figure 4 Coronaviridae) (Malone et al., 2022). Subgenomic negative-sense RNAs are the templates for the “continuous” RNA synthesis of positive-sense subgenomic mRNAs (Sawicki and Sawicki 2005).
Regulation of template switching is complex, and may involve viral RNA, viral proteins and host factors. Nucleocapsid phosphorylation by host glycogen synthase kinase-3 (GSK-3) contributes to template switching, which allows the recruitment of the RNA helicase DDX1 to the phosphorylated-N-containing complex and hence promotes TRS read through of body TRSs to produce longer RNA species in the orthocoronavirus MHV (Wu et al., 2014). In the orthocoronavirus SARS-CoV-2, three DDX DEAD-box RNA helicases are required for viral RNA synthesis, and a fourth restricts virus growth (Ariumi 2022). However, a recent study has shown that the channel formed by the Ubl1 domains of nsp3 monomers on the cytoplasmic surface of the double-membrane vesicle (DMV) is too narrow for passage of viral nucleocapsid or free N protein dimers, suggesting that the N proteins are located on the opposite side of the DMV where viral RNA synthesis takes place (Koetzner et al., 2022). Hence, N protein may not be able to interact directly with the enzymatic components of RNA synthesis. It is proposed that N protein is involved in RNA synthesis by allowing gRNA to enter the DMV while its RNA chaperone activity may play a role in maintaining the long gRNA in a disentangled state which can thread through the channel pore (Zuniga et al., 2010, Koetzner et al., 2022).
Mutagenic studies have shown that exonuclease activity of nsp14 is required to promote fidelity by mediating proofreading during genome replication (Eckerle et al., 2007). The fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants (Eckerle et al., 2007, Eckerle et al., 2010). Infidelity of SARS-CoV nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing (Eckerle et al., 2010). An additional nsp14 function in discontinuous synthesis of subgenomic RNAs has been reported (Ogando et al., 2020, Gribble et al., 2021). It remains to be confirmed that coronavirus-encoded papain-like proteases are essential for subgenomic RNA synthesis, as has been shown for members of the nidoviral family Arteriviridae (Tijms et al., 2001).
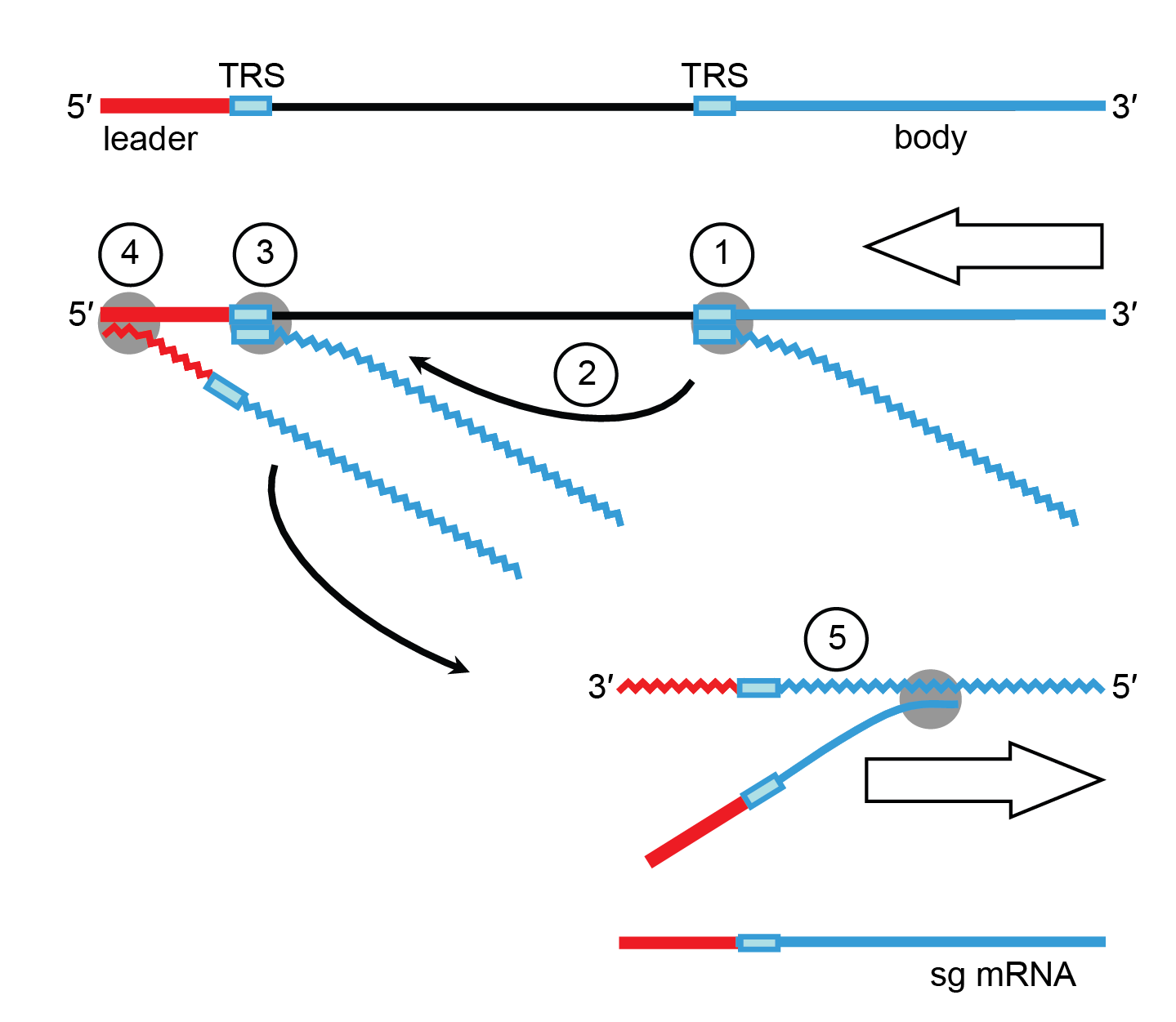 |
| Figure 4.Coronaviridae. Orthocoronavirus mRNA synthesis: the discontinuous 3′-extension model. Negative-sense RNA synthesis initiates at the 3′-end of the genome and proceeds until a TRS is copied (1). The nascent negative-sense RNA may then be transferred to the 5′-end of the genome (2). Base complementarity allows the negative-sense RNA to anneal to the leader TRS (3) after which RNA synthesis resumes and body (in blue) and leader sequences (in red) become fused (4). The chimeric sg negative-sense RNA in turn serves as a template for “continuous” synthesis of sg mRNAs (5). |
A predicted −1 ribosomal frameshift signal is found in the letovirus genome (Bukhari et al., 2018) comprising a slippery sequence followed immediately by a UAA stop codon at the end of polyprotein 1a (Bukhari et al., 2018). A predicted stem-loop conformation (potentially an RNA pseudoknot) is also found in the letovirus genome, suggesting possible ribosomal frameshifting event but further biological characterization is needed (Bukhari et al., 2018).
Information about the pironavirus genome is limited to a single assembly of 36 652 nucleotides.
Biology
The family Coronaviridae includes three sub-families, Orthocoronavirinae, Letovirinae and Pitovirinae. Orthocoronaviruses infect a wide range of animals, including birds and mammals, while letoviruses and pironaviruses are only reported to infect frogs and fish respectively (Bukhari et al., 2018, Mordecai et al., 2019). The transmission routes for orthocoronaviruses are mainly through contact of contaminated surfaces or objects, fomites, respiratory aerosols/droplets and fecal–oral means, but are unknown for letoviruses and pironaviruses (Drosten et al., 2014, Richard et al., 2020, Smith et al., 2020). Members of the subfamily Orthocoronavirinae display diverse receptor usage (Table 3.Coronaviridae) and pathogenicity in different hosts, ranging from infections being asymptomatic to causing death. Some members of the subfamily have been responsible for well-known human epidemics or pandemics such as SARS, MERS and COVID-19 (Ksiazek et al., 2003, Peiris et al., 2003, Zaki et al., 2012, Zhu et al., 2020).
Table 3.Coronaviridae Orthocoronavirus primary attachment factors and receptors*
| Virus genus/species | Characteristics of members of the species | Reference | ||
| Host | Attachment factor | Main receptor | ||
| Genus Alphacoronavirus | ||||
| Alphacoronavirus suis | Dog, cat and pig | Sialic acid | APN | (Delmas et al., 1992, Sanchez et al., 2019) |
| Alphacoronavirus chicagoense | Human | - | APN | (Yeager et al., 1992) |
| Alphacoronavirus amsterdamense | Human | - | ACE2 | (Hofmann et al., 2005, Pohlmann et al., 2006, Smith et al., 2006, Mathewson et al., 2008, Dijkman et al., 2012a) |
| Alphacoronavirus porci | Pig | Sialic acid | APN | (Liu et al., 2015) |
| Genus Betacoronavirus | ||||
| Betacoronavirus gravedinis | Human, cow, horse and pig | - | 9-O-Ac Sia | (Hulswit et al., 2019, Tortorici et al., 2019) |
| Betacoronavirus hongkongense | Human | - | 9-O-Ac Sia | (Huang et al., 2015, Hulswit et al., 2019) |
| Betacoronavirus muris** | Mouse | 4-O- or 9-O-Ac Sia | CEACAM1a | (Williams et al., 1991, Langereis et al., 2010, Langereis et al., 2012) |
| Betacoronavirus pandemicum*** | Human and diverse mammals | - | ACE2 | (Li et al., 2005a, Mathewson et al., 2008, Ge et al., 2013, Letko et al., 2020) |
| Betacoronavirus cameli**** | Human and camel | GRP78, α2,3-sialic acid | DPP4 | (Raj et al., 2013, Li et al., 2017, Chu et al., 2018, Widagdo et al., 2019) |
| Betacoronavirus tylonycteridis | Bat | - | DPP4 | (Yang et al., 2014b, Lau et al., 2021) |
| Betacoronavirus rousetti | Bat | GRP78 | - | (Chu et al., 2018) |
| Deltacoronavirus | ||||
| Deltacoronavirus suis | Pig | Sialic acid | APN | (Wang et al., 2018, Zhu et al., 2018) |
| Gammacoronavirus | ||||
| Gammacoronavirus galli | Poultry | Sialic acid | - | (Winter et al., 2006) |
Abbreviations: APN, aminopeptidase N; ACE2, angiotensin-converting enzyme 2; CEACAM1a, carcinoembryonic antigen adhesion molecule 1; DPP4, dipeptidyl peptidase-4; GRP78, 78 kDa glucose-regulated protein.
*Attachment factors and receptors are virus strain-specific.
** Murine coronaviruses occur in two types that use either 4- or 9-O-acetylated sialic acid (O-Ac Sia) as primary attachment factor and CEACAM1a as main receptor.
*** Only certain strains can utilize ACE2 as receptor.
**** Only certain strains can utilize DPP4 as receptor.
Orthocoronaviruses can infect a wide range of birds and mammals and include several pathogens of clinical, veterinary and economic importance (Woo et al., 2009a). Transmission occurs via fomites, respiratory or fecal–oral routes; vector-borne transmission has not been described to date (Zhang et al., 2020, Guo et al., 2021). As orthocoronaviruses primarily target epithelial cells, they are generally associated with gastrointestinal and respiratory infections that may be acute or become chronic with prolonged shedding of virus (Sims et al., 2005, van Kampen et al., 2021). In general, these infections are mild and often asymptomatic. Some orthocoronaviruses, however, cause severe, even lethal disease (Rockx et al., 2020). Murine coronaviruses in the genus Betacoronavirus cause hepatitis and severe neurologic disease, resulting in encephalitis, so providing a rodent model for the study of the neuropathogenesis of human multiple sclerosis (Fleming et al., 1987). Some members of the species Alphacoronavirus suis cause fatal immune-mediated systemic infections in their respective hosts, presumably through the infection of cells of the macrophage/monocyte lineage, with widespread inflammatory lesions in multiple organs (Haake et al., 2020).
Nine coronaviruses that infect humans have been identified so far, and four of them, human coronaviruses (HCoV)-OC43, HCoV-229E, HCoV-NL63 and HCoV-HKU1, mostly cause common colds and have long been considered of modest clinical importance (van der Hoek et al., 2004, Woo et al., 2006b, Gaunt et al., 2010, Cui et al., 2019, Vlasova et al., 2022). However, HCoV-OC43 and HCoV-229E may also cause severe lower respiratory tract infections (LRTI) in infants and the elderly, and have been reported to be responsible for about 5% of infant hospitalizations from LRTI, globally (Monto and Lim 1974, Principi et al., 2010). HCoV-NL63 is considered an important cause of croup in children (van der Hoek et al., 2005). Comparatively, HCoV-NL63 and HCoV-OC43 infections occur more frequently in early childhood than HCoV-HKU1 or HCoV-229E (van der Hoek et al., 2010, Dijkman et al., 2012b). Zoonotic transmission was observed for HCoV-OC43 (from cattle to humans) and potentially for HCoV-229E, with a proposed transmission route from bats to humans via camels (Vijgen et al., 2005, Corman et al., 2015, Tao et al., 2017).
In 2002–2003, SARS-CoV caused an epidemic in human populations of a severe pulmonary disease with a mortality rate of 10%. SARS-CoV rapidly spread to four continents, infecting 8096 individuals and claiming 774 deaths before it was contained. Epidemiological evidence indicates that this virus originated in bats, spread to Himalayan palm civets, Chinese ferret badgers and raccoon dogs that were being sold at the wet markets of Guangdong, China. They then entered the human population through handling or consumption of these species (Zhao 2007, Ge et al., 2013). Although SARS has since vanished, the episode does underline the pathogenic potential of coronaviruses and the possibility of novel emerging coronavirus infections arising from cross-species transmissions. In the wake of the SARS epidemic, molecular surveillance and virus discovery studies have yielded evidence for hundreds of novel coronaviruses, among which a study in 2007 identified several viruses in a new genus, Deltacoronavirus infecting various avian hosts (Woo et al., 2007, Woo et al., 2012).
In 2012, a novel coronavirus, Middle East respiratory syndrome-related coronavirus (MERS-CoV), caused another epidemic in human populations with a mortality rate of 35% (Zaki et al., 2012). This virus emerged in the Middle East and spread to other regions and countries, leading to isolated infections and occasionally outbreaks of moderate size in hospitals (Drosten et al., 2015, Hui et al., 2015, Kim et al., 2017). Epidemiological studies indicated a polyphyletic origin of MERS-CoV (arising from multiple ancestral sources), suggesting multiple cross-species jumping events from dromedaries (Sabir et al., 2016). The ultimate host origin of MERS-CoV is also believed to be bats (Annan et al., 2013, Wang et al., 2014c, Yang et al., 2014a, Yang et al., 2014b). Unlike SARS, the MERS epidemic is still on-going in the Middle East region.
In late 2019, another outbreak of pneumonia, named COVID-19, emerged from Wuhan, China and spread rapidly to every corner of the world, causing millions of deaths. The cause of this pandemic is a coronavirus closely related to SARS-CoV, named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses 2020). The high mutation rate of orthocoronaviruses, the global scale of the pandemic as well as the involvement of immunologically compromised patients during infections have led to the rapid emergence of novel variants of concern. In 2022, the omicron strain, with higher infectivity but lower pathogenicity compared to the original strain, led to another wave of SARS-CoV-2 infections globally (Nyberg et al., 2022). So far, no definite evidence for the involvement of specific intermediate animal hosts in the origin of SARS-CoV-2 has been identified even though several plausible candidates have been proposed (V'Kovski et al., 2021, Wacharapluesadee et al., 2021, Worobey et al., 2022).
Bats harbor an exceptionally wide diversity of orthocoronaviruses and have been proposed to play a vital role in orthocoronavirus ecology and evolution, maybe even as the original hosts from which many if not all alphacornavirus and betacoronavirus lineages derive (Tang et al., 2006, Woo et al., 2006a, Lin et al., 2017, Banerjee et al., 2019). Bat population densities and their roosting and migration habits would all favor such a role and it is possible that more detailed surveillance of bats may identify precursors of many human coronaviruses. Equally, surveillance of other potential host species is needed before final conclusions can be drawn.
The only known member of the subfamily Letovirinae, is Microhyla letovirus (MLeV), which was detected in ornamented pygmy frogs (Microhyla fissipes) (Bukhari et al., 2018). Mapping single sequence reads onto the viral genome suggests a strong host age-dependence of MLeV detection. The number of fragments per kb of transcript per million mapped reads decreased by seven-fold from host pre-metamorphosis to metamorphic climax and it further decreased by fourteen-fold from host metamorphic climax to completion of metamorphosis (Bukhari et al., 2018).
Pacific salmon nidovirus (PsNV) (species: Alphapironavirus salmonis), the only member of the subfamily Pitovirinae, is found in salmon (Mordecai et al., 2019). In hatchery fish, infection by PsNV is primarily localized to gill tissue (Mordecai et al., 2019). It proliferates while fish are undergoing smoltification, a biological process by which the gill tissue undergoes cellular reconfiguration to prepare for salt water (Mordecai et al., 2019). Branchial proliferation with unknown cause is also reported in some farmed salmon infected with PsNV (Mordecai et al., 2019).
Antigenicity
For orthocoronaviruses, cross-reactivity is found only among members of closely-related species within the same genus (Wang et al., 2021, Reincke et al., 2022). Several viral proteins, including S, M, N and HE are immunogenic or relevant to serodiagnosis (Huang et al., 2004, Thompson et al., 2020). The S and HE proteins are highly diverse, suggesting extensive antigenic drift through genetic recombination or mutation (Lau et al., 2012).
The S protein is the major inducer of virus-neutralizing antibodies, these being elicited mainly by epitopes in the amino-terminal half of the molecule (Du et al., 2009, Mou et al., 2013, Kim et al., 2014). The surface-exposed amino-terminus of the MHV M protein induces antibodies that neutralize virus infectivity in the presence of complement, while the HE protein of embecoviruses induces antibodies that prevent virions binding to O-acetylated sialic acids or inhibit sialate-O-acetylesterase activity (Fleming et al., 1989, Lin et al., 2001). The N protein is a dominant antigen during natural infections, and while N-specific antibodies may provide little immune protection, they are of serodiagnostic relevance (Tan et al., 2004, Edridge et al., 2020).
There is also evidence for antigenic shift through RNA recombination in coronaviruses as there are several examples of intra- and possibly inter-species exchange of coding sequences of S (for members of the species Gammacoronavirus galli, Betacoronavirus muris and Alphacoronavirus suis) and HE ectodomains (for members of the species Betacoronavirus muris), sometimes with as yet unidentified coronaviruses serving as donors (Su et al., 2016). Studies performed with murine and feline coronaviruses indicate that both structural and non-structural (replicase) proteins are recognized by CD4+ and CD8+ T cells (Williamson and Stohlman 1990, de Groot-Mijnes et al., 2005).
Similar to other coronaviruses, the S1 subunits of the members from the sarbecovirus subgenus are more diverse than the S2 subunits. Only some of the members, including SARS-CoV and SARS-CoV-2, utilize ACE2 as the receptor for host cell entry. Animal experiments show that mice immunized with SARS-CoV S can produce neutralizing antibodies against SARS-CoV-2 S, suggesting that immunity against one member of the subgenus Sarbecovirus may offer cross protection against other viruses in the subgenus (Walls et al., 2020). The S2 subunits of the S glycoprotein in SARS-CoV and SARS-CoV-2 are about 88% identical in amino acid sequence. The conformational conserved fusion regions suggest the antibodies targeting this functional motif might have the ability to cross-react and neutralize both viral strains (Walls et al., 2020).
There is evidence that SARS-CoV-2 variants emerging during the pandemic carry different S amino acid mutations that affect host receptor binding affinity as well as the efficacy of antibody neutralization by post vaccination serum. Studies also hypothesize that treatment of immunocompromised individuals with convalescent plasma and monoclonal antibodies might have led to the emergence of heavily mutated variants like B.1.1.7 and B.1.351 by exerting selective pressure on the S protein, especially at the receptor binding motif and N-terminal domain (NTD) (Harvey et al., 2021).
The extent of serologic cross-reactivity between orthocoronaviruses, letoviruses and pironaviruses is unknown.
Subfamily, genus, subgenus and species demarcation criteria
The taxonomic framework is based upon viruses for which a complete genome sequence is available or else a nearly complete genome that includes the 3CLpro, NiRAN, RdRP, ZBD and HEL1 domains (Ziebuhr et al., 2018, Gorbalenya et al., 2021). Genome sequences are analyzed using the computational framework DEmARC (DivErsity pArtitioning by hieRarchical Clustering), an approach which has been applied to the taxonomy of all nidoviruses (Lauber and Gorbalenya 2012). The analysis involves profiles of multiple sequence alignments (MSA) of 3CLpro, NiRAN, RdRP, ZBD and HEL1 domains made using Bayesian and Maximum-likelihood phylogenetic trees, with profiles of the clustering cost (CC) function produced for weighted hierarchical clusterings of pairwise patristic distances (PPD). The family, subfamily, genus, subgenus and species demarcation thresholds are set as a range of PUD values (% of different amino acid residues in compared proteins, Table 4 Coronaviridae), which are deduced from PPD values for which the number of taxa clusters remained constant.
Table 4 Coronaviridae Demarcation thresholds for five ranks of the order Nidovirales for vertebrate viruses (Ziebuhr et al., 2018, Gorbalenya et al., 2021)
| Rank | Number of taxa | Threshold | Support | |
| Year 2021 | PPD1 | PUD2 | CC3 | |
| Family | 7 | 2.580-3.468 | 0.606-0.681 | 0 |
| Subfamily | 14 | 1.472-1.757 | 0.468-0.510 | 0.005025 |
| Genus | 25 | 0.873-0.909 | 0.351-0.360 | 0.007678 |
| Subgenus | 54 | 0.200-0.221 | 0.132-0.142 | 0.005541 |
| Species | 86 | 0.095 | 0.075 | 0.010339 |
1Demarcation threshold depicted as a range of PPD values for which number of clusters (taxa) remained constant. PPD values account for repeated replacements of amino acid residues and may exceed 1.
2Demarcation threshold depicted as a range of PUD values, deduced from PPD values for which the number of clusters (taxa) remained constant. PUD values were calculated as the % of different residues in the compared proteins.
3CC value of the respective PPD threshold. It was selected for each rank to preserve the already established taxa at the respective rank. For the family and subfamily ranks, the depicted CC corresponds also to local minima of the CC distribution indicating that these selections are also the best possible. For the species, subgenus and genus ranks, the selected CC are in the vicinity of local minima. If the actual local minima had been selected, one or two established taxa would have been revised at each of these ranks.
Corresponding taxonomic proposal(s): 2021.005S.R.Nidovirales
Derivation of names
Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus: genus names refer to phylogroups 1 to 4 respectively in the subfamily Orthocoronavirinae, the prefixes corresponding to the first four Greek letters.
Coronaviridae: from Latin corona, meaning “halo” which refers to the characteristic appearance of virion surface projections that create an image reminiscent of the solar corona (Almeida et al., 1968).
Letovirinae: from Leto, in Greek mythology the daughter of the titans Coeus and Phoebe, who turned some inhospitable peasants into frogs; the name refers to frogs as the host source of this virus (Bukhari et al., 2018).
Orthocoronavirinae: from Greek ὀρθός (orthós) meaning straight; Coronavirinae is derived from the established name of this group of viruses (coronaviruses).
Pironavirus: origin obscure. Names of species and subgenus taxa in the single new genus allude to the names of the first reported virus in those taxa.
Pitovirinae: origin obscure.
Subgenus names are generally formed from a unique part derived from a host or virus name, followed by the first one or two letters of the genus name (family name for Milecovirus)and ending with ‘covirus’ (‘ovirus’ for Samovirus) (de Groot et al., 2008, Brinton et al., 2018, Gorbalenya et al., 2021).
Relationships within the family
Members of the family Coronaviridae have been assigned to three sub-families, Orthocoronavirinae, Letovirinae and Pitovirinae, on the basis of rooted phylogeny and pair-wise comparisons of Nidovirales-wide conserved replicase domains. Within the Orthocoronavirinae, four well-separated monophyletic clusters can be distinguished using a rooted maximum-likelihood tree generated from amino acid sequence alignments of (a) RdRP and (b) helicase domains. Microhyla letovirus 1 was used as an outgroup. These clusters correspond to the genera Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus (Figure 5A Coronaviridae and Figure 5B Coronaviridae). Orthocoronaviruses have been detected in diverse mammalian and avian animals. Two divergent viruses detected respectively in frogs and fish are classified in single species in the sub-families Letovirinae and Pitovirinae.
 |
| Figure 5 Coronaviridae. Phylogenetic relationships among members of the family Coronaviridae. A rooted maximum-likelihood tree was generated from amino acid sequence alignments of (A) RdRP and (B) helicase domains. Microhyla letovirus 1 was used as an outgroup. The tree reveals four main monophyletic clusters corresponding to the genera Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus within the subfamily Orthocoronavirinae. The assignment of viruses to taxa is based upon previous taxonomic proposals (Ziebuhr et al., 2018, Gorbalenya et al., 2021). |
Relationships with other taxa
Based on the ML phylogenetic trees constructed using multiple sequence alignments of five concatenated domains in the replicase region (3CLpro, NiRAN, RdRP, ZBD and HEL1) of members of the order Nidovirales, members of the family Coronaviridae are most closely related to members of the family Tobaniviridae that have vertebrate hosts and were previously included in the family Coronaviridae (Zhou et al., 2021). The family Coronaviridae consistently forms a monophyletic cluster that is separate from other nidoviral families including the Arteriviridae, Tobaniviridae and Roniviridae. Similarities in genomic organization, sequences of conserved replicase proteins, structural proteins and the subgenomic mRNA transcription strategy suggest a close relationship between members of the family Coronaviridae with other members of the order Nidovirales.
Related, unclassified viruses
There are recent reports of coronavirus-like sequences from two more vertebrate classes; a divergent lineage in jawless fish (Miller et al., 2021) and a partial orthocoronavirus-like genome from a reptile (Shi et al., 2018), both of which have yet to be formally reviewed by the ICTV.