

Family: Circoviridae
Genus: Cyclovirus
Distinguishing features
Cyclovirus genomes contain two major ORFs encoding the replication-associated protein (Rep) and capsid protein (Cp). . In contrast to members of the genus Circovirus, cycloviruses have the putative ori on the Cp-encoding strand (Rosario et al., 2012). In addition, cyclovirus genomes have an intergenic region (IR) between the 5′-end of major ORFs; however, the IR between the 3′-ends of these ORFs is smaller than that of genomes representing the genus Circovirus or may be absent (Li et al., 2010, Delwart and Li 2012). Introns have been identified within the ORFs of several cyclovirus genomes, while none have been reported for circoviruses.
Virion
Morphology
Unknown.
Physicochemical and physical properties
Unknown.
Nucleic acid
Cycloviruses exhibit small, circular ssDNA genomes ranging from 1.7 to 1.9 kb.
Proteins
Unknown.
Lipids
Unknown.
Carbohydrates
Unknown.
Genome organization and replication
Cyclovirus genomes contain two major ORFs encoding the Rep and Cp (Figure 1. Cyclovirus) (Rosario et al., 2017). Although the virion strand has not be experimentally demonstrated for cycloviruses, knowledge from other eukaryotic Rep-encoding ssDNA viruses suggests that the strand containing the nonanucleotide motif is encapsidated (Rosario et al., 2012). Since the putative cyclovirus ori and predicted Rep share conserved features with members of the genus Circovirus, cycloviruses are also thought to replicate through rolling circle replication (RCR). Similar to circoviruses, the putative cyclovirus ori contains the canonical nonanucleotide motif ‘NAGTATTAC’ located at the apex of a potential stem-loop structure found between the 5ʹ-ends of major ORFs. In addition, the Rep contains endonuclease and helicase domains with RCR and SF3 motifs similar to those identified in circovirus Reps. These conserved motifs include RCR motifs I [FT(L/W)NN], II [(P/x)HLQG] and III [Y(C/l)(S/x)K] and SF3 helicase motifs Walker-A [G(P/x)(P/t)(G/x)xGKS], Walker-B [uuDDF], and motif C [uTS(N/e)], where ‘‘x’’ represents any type of residue and ‘‘u’’ represents a hydrophobic amino acid (i.e., F, I, L, V, M). Moreover, putative Rep-binding domains characterized by iterative sequences near the stem-loop structure have been identified for some cyclovirus genomes (Dayaram et al., 2013).
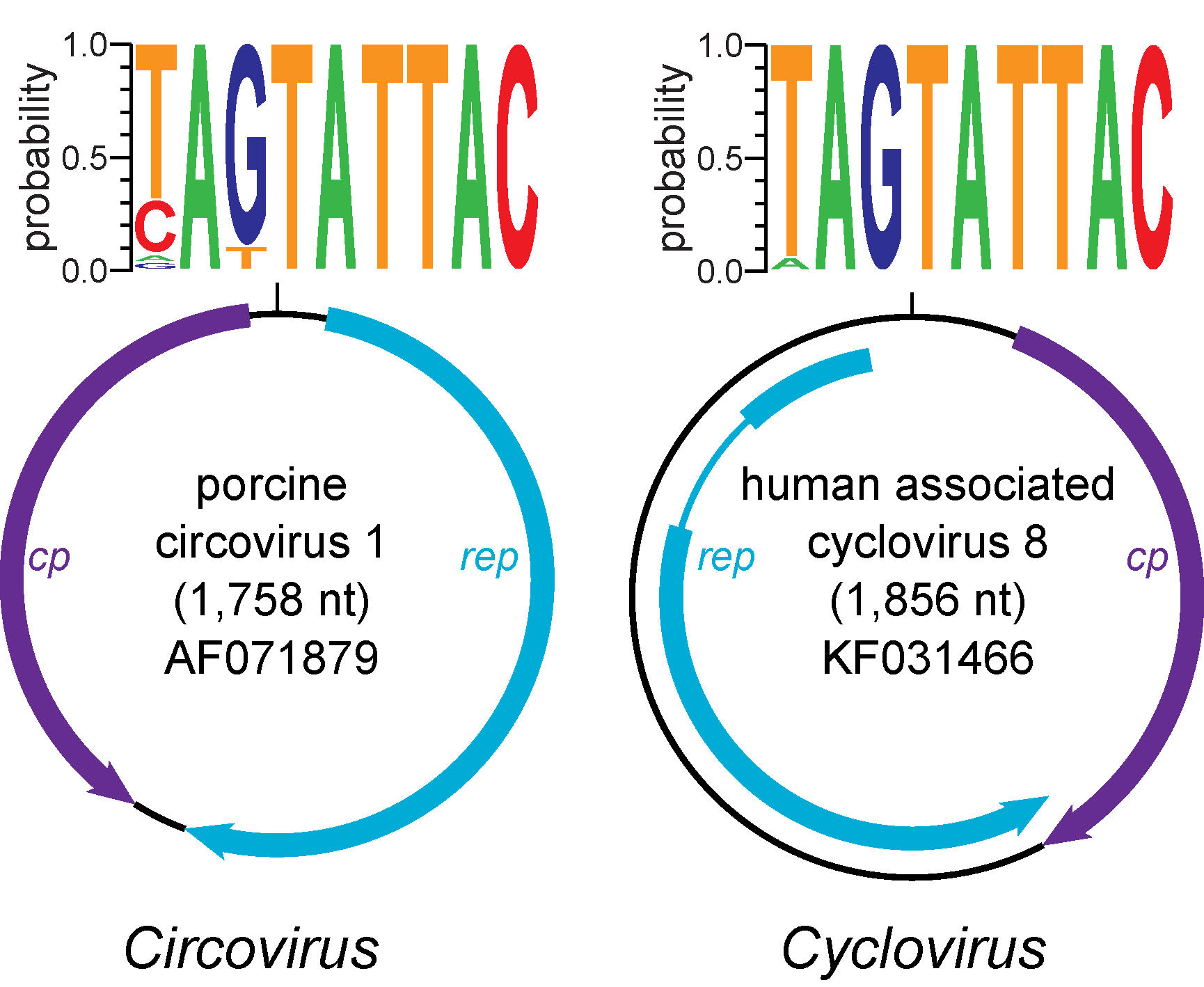 |
| Figure 1. Cyclovirus. Genome schematics illustrate the major open reading frames (ORFs) characteristic of members of the Circoviridae family. Members of the family Circoviridae, including the Circovirus and Cyclovirus genera, have two major ORFs encoding replication-associated (Rep) and capsid (Cp) proteins as well as a conserved nonanucleotide motif marking the origin of replication. The nonanucleotide motif sequence is depicted through sequence probability logos generated in Weblogo 3 (Crooks et al., 2004). Note that the orientation of major ORFs relative to the nonanucleotide motif differs between genomes representing the Circovirus and Cyclovirus genera. The rep of members of the Cyclovirus type species, Human associated cyclovirus 8 is interrupted by an intron. Although the presence of introns has been observed in various cyclovirus genomes, this has not been reported for circoviruses. Figure reprinted with permission of Springer from Archives of Virology, “Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus”, doi: 10.1007/s00705-017-3247, Rosario, K., Breitbart, M., Harrach, B., Segales, J., Dewart, E., Biagini, P., Varsani, A. © Springer-Verlag Wien 2017 (Rosario et al., 2017). |
Biology
Cyclovirus genomes were originally discovered in primate stool samples and meat products from a diverse range of animals (camels, chickens, cows, goats, sheep) (Li et al., 2010, Delwart and Li 2012). However, cyclovirus genomes have now been reported from various types of human samples other than faeces, including cerebrospinal fluid, blood serum, and respiratory secretions (Smits et al., 2013, Tan le et al., 2013, Phan et al., 2014, Phan et al., 2015). Additionally, cycloviruses have been detected in samples from a wide range of mammalian and insect species (Rosario et al., 2017). Since genomes belonging to the genus Cyclovirus have only been identified through sequence-based analyses (i.e., degenerate PCR and metagenomic sequencing), definitive hosts and biology are largely unknown.
Derivation of names
Cyclo: from cyclo, latinised form of kyklos in Greek, meaning a circle, wheel or ring
Species demarcation criteria
The species demarcation threshold is 80% genome-wide nucleotide sequence identity based on pairwise identity distribution analysis (Figure 2. Cyclovirus) (Rosario et al., 2017).
 |
| Figure 2. Cyclovirus. Distribution of pairwise identities among members of the genus Cyclovirus (purple bars; left) and the genus Circovirus (blue bars; right). Plots reflect pairwise identities based on calculations for the complete genome sequences (top) as well as the replication-associated (rep; middle) and capsid (cp; bottom) genes. All pairwise identities were calculated using the Sequence Demarcation Tool version 1.2 (Muhire et al., 2014) with the MUSCLE alignment algorithm (Edgar 2004). Figure reprinted with permission of Springer from Archives of Virology, “Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus”, doi: 10.1007/s00705-017-3247, Rosario, K., Breitbart, M., Harrach, B., Segales, J., Dewart, E., Biagini, P., Varsani, A. © Springer-Verlag Wien 2017 (Rosario et al., 2017). |