
Family: Orthoherpesviridae
Subfamily: Alphaherpesvirinae
Distinguishing features
The predicted amino acid sequences of members form a distinct lineage within the family (Figure 1.Alphaherpesvirinae). A region of the genome comprising the unique short sequence (US) and flanking internal and terminal inverted repeats (IRL/TRL and IRS/TRS) contains genes that are specific to the subfamily. Most members have a class 3 or 4 genome architecture (Figure 2. Orthoherpesviridae and Figure 3. Orthoherpesviridae). Many members of the family with the highest G+C content belong to this subfamily. Splicing is rarer than in members of the other subfamilies.
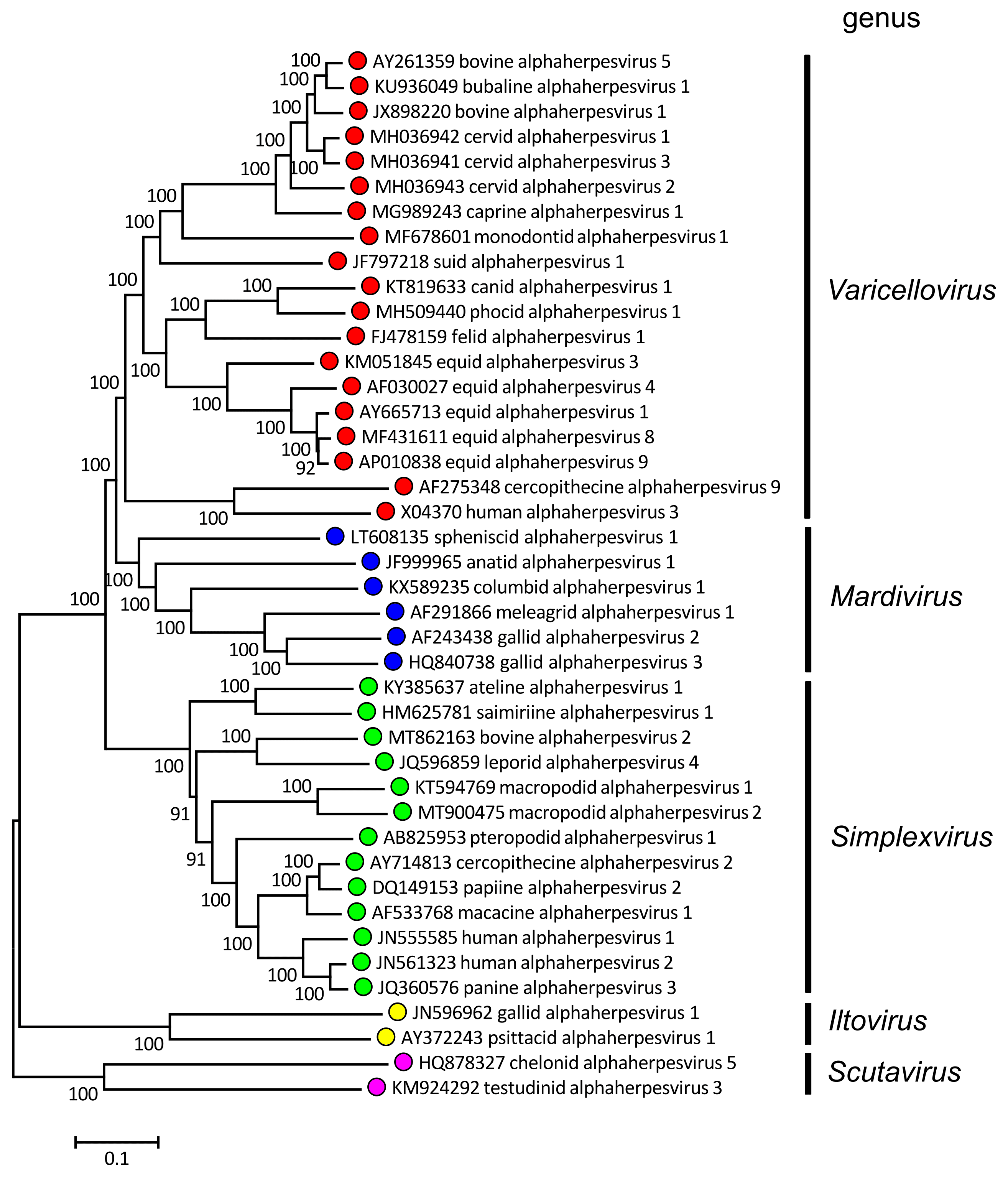 |
| Figure 1.Alphaherpesvirinae. Phylogenetic relationships within the subfamily Alphaherpesvirinae. Alignments of predicted amino sequences (Resource 1) were performed using MUSCLE (Edgar 2004). Phylogenetic trees were computed from the alignments (Resources 2–4) using the neighbor-joining method (Saitou and Nei 1987) with 500 bootstrap replicates (Felsenstein 1985). Evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling 1965) and are in the units of the number of amino acid substitutions per site. The alignments and default midpoint-rooted trees were produced using MEGA 7 and MEGA X (Kumar et al., 2016, Kumar et al., 2018). The viruses are represented by systematic virus names. Resource 1 provides correspondences to virus names where these differ from systematic names. Coloured circles denote individual genera within the subfamily. This phylogenetic tree and corresponding sequence alignment are available to download from the Resources page. |
Members have been found in a wide range of mammalian hosts and also in avian and reptilian hosts. They have a broad host cell range in cell culture, with a relatively rapid cytolytic productive cycle. Latent infection is established in neurons in vivo, although other sites have also been reported.
Genus demarcation criteria
See discussion under family description.