

Family: Orthoherpesviridae
Derek Gatherer, Daniel P. Depledge, Carol A. Hartley, Moriah L. Szpara, Paola K. Vaz, Mária Benkő, Curtis R. Brandt, Neil A. Bryant, Akbar Dastjerdi, Andor Doszpoly, Ursula A. Gompels, Naoki Inoue, Keith W. Jarosinski, Rajeev Kaul, Vincent Lacoste, Peter Norberg, Francesco C. Origgi, Richard J. Orton, Philip E. Pellett, D. Scott Schmid, Stephen J. Spatz, James P. Stewart, Jakob Trimpert, Thomas B. Waltzek and Andrew J. Davison
Summary
Members of the family Orthoherpesviridae have enveloped, spherical virions with characteristic complex structures consisting of symmetrical and non-symmetrical components (Table 1.Orthoherpesviridae). The linear, double-stranded DNA genomes of 125–241 kbp contain 70–170 genes, of which 43 have been inherited from an ancestral herpesvirus. Orthoherpesviruses have generally coevolved with their hosts and are highly adapted to them, and are likely to be associated with most mammalian, avian and reptilian species. Following primary infection, they are able to establish life-long latent infection, during which there is limited viral gene expression. Severe disease is usually observed only in the foetus, the very young, the immunocompromised or following infection of an alternative host.
Table 1.Orthoherpesviridae. Characteristics of members of the family Orthoherpesviridae
| Characteristic | Description |
| Example | herpes simplex virus 1 (JN555585), species Simplexvirus humanalpha1, genus Simplexvirus, subfamily Alphaherpesvirinae |
| Virion | Spherical (150–200 nm) particles consisting of a tightly condensed DNA core, an icosahedral capsid, a tegument (matrix) and a glycoprotein-containing lipid envelope |
| Genome | 125–241 kbp of linear dsDNA |
| Replication | Infection has lytic and latent phases; transcription occurs in the nucleus by a kinetic cascade; DNA replicates by a rolling-circle mechanism to generate concatemers, from which genomes are cleaved and packaged into preformed capsids; virions mature in the cytoplasm and leave the cell by exocytosis |
| Translation | Occurs from capped, polyadenylated mRNAs, some of which are spliced |
| Host range | Mammals, birds and reptiles |
| Taxonomy | Realm Duplodnaviria, kingdom Heunggongvirae, phylum Peploviricota, class Herviviricetes, order Herpesvirales, including 3 subfamilies, 17 genera and 118 species |
Virion
Morphology
Virions have a characteristic, complex structure consisting of symmetrical and non-symmetrical components. Mature virions of herpes simplex virus "type" 1 (HSV1 in the species Simplexvirus humanalpha1) are pleomorphic and have an average diameter of 186 nm at the base of the envelope and approximately 225 nm when the glycoprotein spikes are included (Grünewald et al., 2003). The virion comprises a core, capsid, tegument (matrix) and envelope (Figure 1.Orthoherpesviridae).
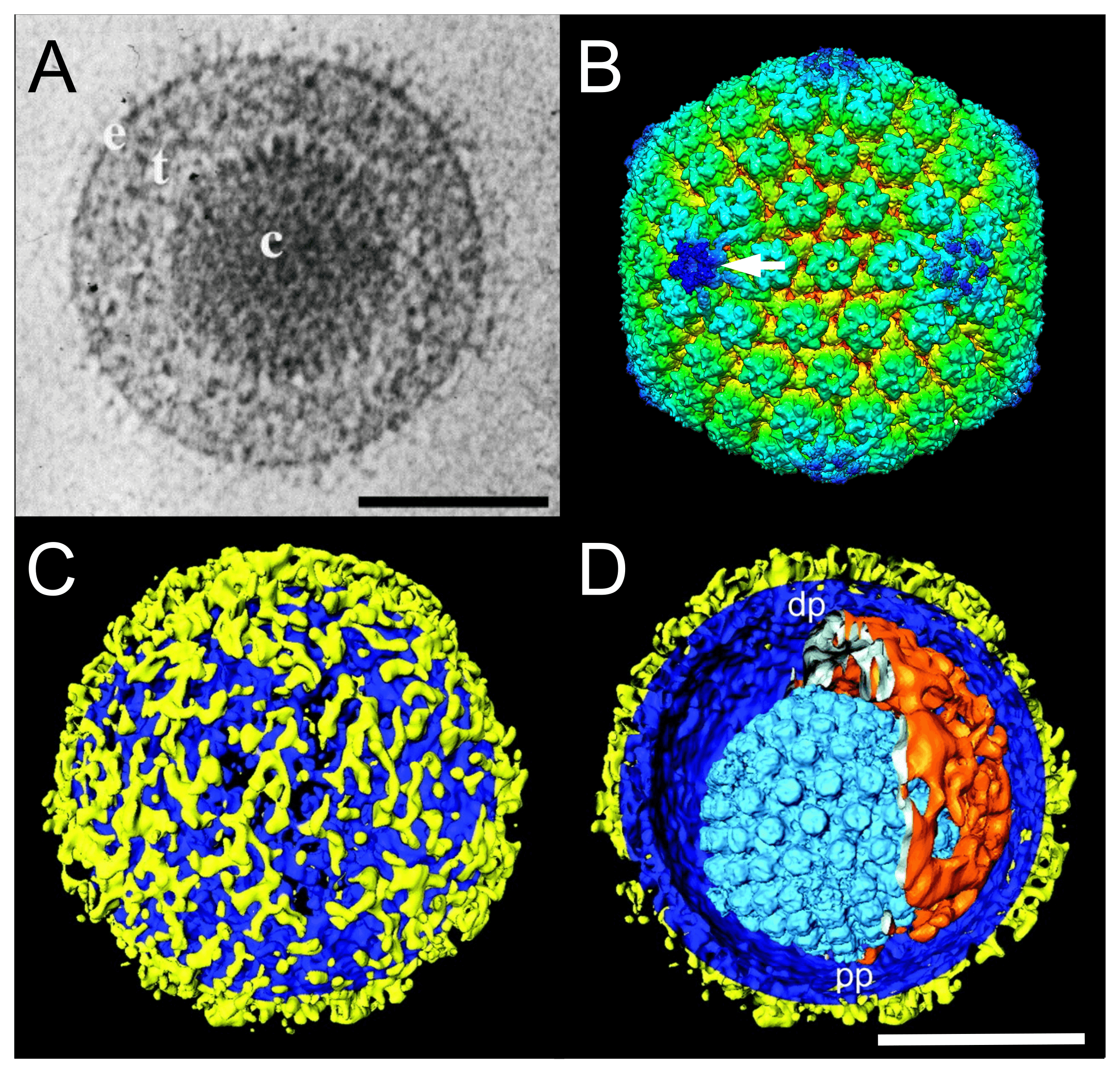 |
| Figure 1.Orthoherpesviridae. Virion and capsid structure. (A) Cryo-electron microscopy image of a single herpes simplex virus 1 (HSV1) virion. The capsid (c) is embedded in the tegument (t) and enclosed by a lipid envelope containing glycoproteins (e). Scale bar, 100 nm. From (McGeoch et al., 2006) with permission. (B) Three-dimensional cryo-electron microscopy image reconstruction of a single HSV1 capsid indicating the unique portal-vertex by a white arrow. From (McElwee et al., 2018). (C and D) Segmented surface rendering of a single HSV1 virion tomogram after denoising. From (Grünewald et al., 2003) with permission. (C) Outer surface showing the distribution of glycoprotein spikes (yellow) protruding from the membrane (blue). (D) Cutaway view of the virion interior, showing the capsid (light blue) and the asymmetric tegument cap (orange) inside the envelope (blue and yellow). pp, proximal pole; dp, distal pole. Scale bar, 100 nm. |
The core consists of the viral genome packaged as a single, linear, double-stranded DNA molecule into a preformed capsid. The DNA exists under pressure in a liquid-crystalline array that fills the entire internal volume of the capsid (Booy et al., 1991).
The mature capsid is a T=16 icosahedron. In HSV1 virions, the 16 nm-thick protein shell has an external diameter of 125 nm. The 11 pentons and 150 hexons (a total of 161 capsomers) are composed primarily of five and six copies, respectively, of the major capsid protein VP5 (encoded by gene UL19) and are joined by masses, termed triplexes, that are made of two smaller proteins VP23 and VP19C (encoded by genes UL18 and UL38, respectively) in a 2:1 ratio (Homa and Brown 1997, Chiu and Rixon 2002). The twelfth pentonal position is occupied by a ring-like portal structure consisting of 12 copies of the capsid portal protein (encoded by gene UL6) that serves as the channel through which the genome is inserted into the capsid (McElwee et al., 2018). The presence of capsid proteins VP5, VP23, VP19C and the small external protein VP26 (encoded by gene UL35) in multiple conformations has been confirmed by cryo-electron microscopy (Dai and Zhou 2018). Capsids assemble in the infected cell nucleus by nucleation on the portal complex and then assembly of capsid protein complexes around a protein scaffold (encoded by genes UL26 and UL26.5) to form a procapsid in which the subunits are weakly connected. Proteolytic cleavage of the scaffolding proteins by the UL26-encoded protease triggers loss of the scaffold and reorganization of the shell into the characteristic capsid form (Gao et al., 1994).
The tegument is structurally the most complex part of the virion, but remains poorly defined (Kelly et al., 2009). Located between the capsid and the envelope, it contains many viral (and possibly some host) proteins, as well as viral and cellular transcripts (Sciortino et al., 2001, Loret et al., 2008), not all of which are required for virion formation. Individual tegument proteins can vary markedly in abundance. Enveloped tegument structures lacking capsids can assemble in infected cells in cell culture and are released from cells along with virions; these are termed light or L-particles in HSV1 (Szilágyi and Cunningham 1991, McLauchlan and Rixon 1992) and dense bodies and noninfectious enveloped particles in human cytomegalovirus (HCMV in the species Cytomegalovirus humanbeta5) (Talbot and Almeida 1977, Irmiere and Gibson 1983). Electron tomography indicates that there are inner (capsid-associated) and outer (envelope-associated) tegument layers in virions, and that capsids may be situated non-centrally within the envelope to form an asymmetric tegument cap (Grünewald et al., 2003). The capsid-associated tegument complex harbours the proteins encoded by UL17, UL25 and UL36, the latter linking the capsid to the outer tegument and envelope (Dai and Zhou 2018).
The envelope consists of a lipid bilayer that is intimately associated with the outer surface of the tegument and contains several (at least 11 for HSV1) different integral viral glycoproteins that form a network of closely spaced spikes (a mean of 659 per HSV1 virion) of at least three distinct morphologies (Grünewald et al., 2003).
Physicochemical and physical properties
The HSV1 virion has a mass of about 13×10−16 g, of which DNA comprises about 10% (Lampert et al., 1969), and a buoyant density of 1.726 g cm−3 (Goodheart et al., 1968, Kieff et al., 1971). The mass of the full capsid alone is about 7×10−16 g. The buoyant density of virions in CsCl is 1.22–1.28 g cm−3 (Spear and Roizman 1967). Family members vary considerably in stability, but are generally destabilized by desiccation and low pH. Infectivity is destroyed by lipid solvents and detergents (Andrewes and Horstmann 1949, Kaplan and Vatter 1959).
Nucleic acid
The linear, double-stranded DNA molecule in the virion core ranges from 125 kbp (varicella-zoster virus [VZV] in the species Varicellovirus humanalpha3 (Davison and Scott 1986); and simian varicella virus [SVV] in the species Varicellovirus cercopithecinealpha9 (Gray et al., 2001)) to 241 kbp (chimpanzee cytomegalovirus [CCMV] in the species Cytomegalovirus paninebeta2 (Davison et al., 2003)). Members of the subfamily Alphaherpesvirinae, especially those in the genus Varicellovirus, range very widely in nucleotide composition (Honess 1984), from 32% G+C (canine herpesvirus in the species Varicellovirus canidalpha1) to 78% G+C (red deer herpesvirus in the species Varicellovirus cervidalpha1). The genomes examined in sufficient detail have been shown to contain a single nucleotide extension at the 3′-end of each strand, and no terminal protein has been identified (Mocarski and Roizman 1982).
The arrangement of reiterated sequences (direct or inverted repeats at the genome termini or internally) results in several classes of genome architecture, some including orientation isomers resulting from recombination between terminal and internal inverted repeats (Figure 2.Orthoherpesviridae). Class 1 genomes consist of a unique sequence flanked by a direct repeat that may be as short as 31 bp (murine cytomegalovirus in the species Muromegalovirus muridbeta1 (Rawlinson et al., 1996)) or as long as several kbp (human herpesviruses 6A and 6B [HHV6A and HHV6B] in the species Roseolovirus humanbeta6a and Roseolovirus humanbeta6b (Martin et al., 1991, Lindquester and Pellett 1991)). Class 2 genomes also contain a unique sequence, but in this case it is flanked by a variable number of direct repeats at each terminus (e.g. herpesvirus saimiri in the species Rhadinovirus saimiriinegamma2 (Bornkamm et al., 1976); and Kaposi’s sarcoma-associated herpesvirus [KSHV] in the species Rhadinovirus humangamma8 (Russo et al., 1996)). Class 3 genomes contain different elements at each terminus that are also present internally in inverted orientation. The genome is thus divided into two unique regions (one long and one short), which are flanked by inverted repeats. The repeat flanking the long unique sequence is very short (32 bp in equine abortion virus [EHV1] in the species Varicellovirus equidalpha1 (Yalamanchili and O'Callaghan 1990, Telford et al., 1992); and 88 bp in VZV (Davison 1984)) or absent (e.g. pseudorabies virus in the species Varicellovirus suidalpha1 (DeMarchi et al., 1990)). Homologous recombination in replicated concatemeric DNA results in inversion of the two unique regions, and cleavage largely or entirely at one of the two junction regions results in unit length genomes that are one or the other of two isomers differing in the orientation of the short unique region (Davison 1984, DeMarchi et al., 1990). Class 4 genomes have the most complex architecture. Like class 3 genomes, they contain long and short unique regions, but these are both flanked by large inverted repeats. Homologous recombination and cleavage occur with equal probability at either of the two junction regions and result in the formation of four isomers differing in the orientations of the unique sequences, with each isomer equimolar in virion populations (e.g. HSV1 (Sheldrick and Berthelot 1975, Hayward et al., 1975)). In addition, class 4 genomes contain a short terminal direct repeat, which is also present internally in inverse orientation at the junction region. The different isomers of the HSV1 genome appear to be functionally equivalent (Mahiet et al., 2012).
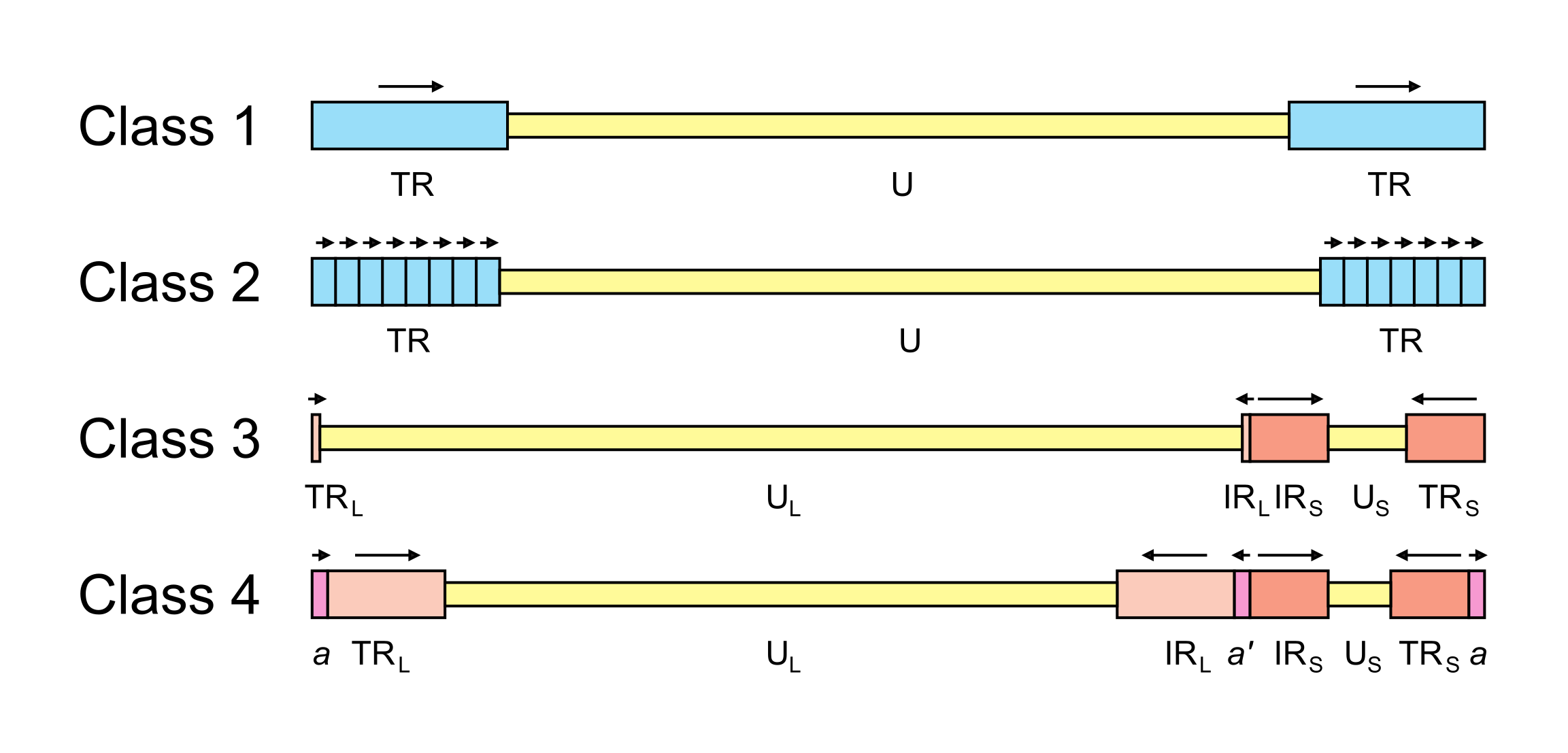 |
| Figure 2.Orthoherpesviridae. Major classes of orthoherpesvirus genome architecture. In this simplified, not-to-scale illustration, unique and repeated sequences are shown in thinner and thicker format, respectively. The nomenclature used to describe these sequences is U (unique), UL (long unique), US (short unique), TR (terminal repeat), TRL (terminal long repeat), IRL (internal long repeat), TRS (terminal short repeat) and IRS (internal short repeat). Class 4 genomes contain a terminal direct repeat (a) that is also present internally in inverse orientation (a’); formally, a is part of both TRL and TRS, and a’ is part of both IRL and IRS. Unique sequences are shaded yellow, direct repeats are shaded blue, inverted repeats are shaded red and a and a’ are shaded pink. The orientations of repeated sequences are shown by arrows. Further details and examples of viruses belonging to each class are provided in the text of this section. |
A few family members contain large repeats within the genome that are unrelated to those at the termini (e.g. Epstein-Barr virus [EBV] in the species Lymphocryptovirus humangamma4 (Rymo and Forsblom 1978)), and a more complex set of structures can be catalogued if these are included. Thus, in the summary of genome architectures depicted in (Krug and Pellett 2021), types A, B, D and E correspond to classes 1, 2, 3 and 4, respectively, in Figure 2.Orthoherpesviridae. Type C corresponds to the EBV genome (essentially a class 2 genome with a large internal direct repeat), and type F corresponds to the genome of tupaia herpesvirus (in the species Quwivirus tupaiidbeta1), which lacks direct or inverted terminal repeats (Bahr and Darai 2001).
Particular genome structures are associated with certain taxa. Thus, class 2 genomes are common in members of the subfamily Gammaherpesvirinae (though not all members have this structure), and class 3 genomes are associated with members of the genus Varicellovirus in the subfamily Alphaherpesvirinae. However, distantly related viruses may have equivalent genome structures that have presumably evolved convergently. For example, HHV6A and HHV6B (subfamily Betaherpesvirinae) and equine herpesvirus 2 (in the species Percavirus equidgamma2, subfamily Gammaherpesvirinae) have class 1 genomes (Telford et al., 1995), and HSV1 (subfamily Alphaherpesvirinae) and HCMV (subfamily Betaherpesvirinae) have class 4 genomes (Weststrate et al., 1980).
Proteins
The protein composition of the mature virion varies greatly among family members. More than 30 different protein species have been identified in HSV1 virions; others likely remain to be discovered. For each protein species, the number of discrete protein or peptide molecules present in a virion ranges from fewer than 50 to almost 1,500 (Heine et al., 1974). The mature capsid is composed of four major and several minor proteins, and the tegument contains at least 15 different proteins (Dai and Zhou 2018), many of which are dispensable in cell culture and are therefore not required for virion morphogenesis. The viral envelope contains at least ten (and in some viruses many more) integral membrane proteins, a subset of which is required for adsorption and penetration of the host cell, where they can also be expressed on the host cell membrane. Host proteins can be present in virions, but their functional significance has not been demonstrated (Loret et al., 2008, Spear and Roizman 1972).
Lipids
The lipid composition of the virion envelope has been reported for a few family members. The HSV1 envelope is enriched in sphingomyelin and phosphatidylserine and resembles the composition of Golgi membranes more closely than that of other cellular membranes (van Genderen et al., 1994). During HCMV infection, there is increased production of very long chain fatty acids, and saturated forms of these lipids are incorporated selectively into the virion envelope (Koyuncu et al., 2013).
Carbohydrates
Virion envelopes contain multiple proteins that may bear N-linked and O-linked glycans. Mature, cell-free virions contain complex glycans, whereas some intracellular virions contain N-linked glycans of the immature high-mannose type (Johnson and Spear 1983). O-linked glycosylation sites have been mapped globally for several orthoherpesviruses by mass spectrometric methods (Bagdonaite et al., 2016).
Genome organization and replication
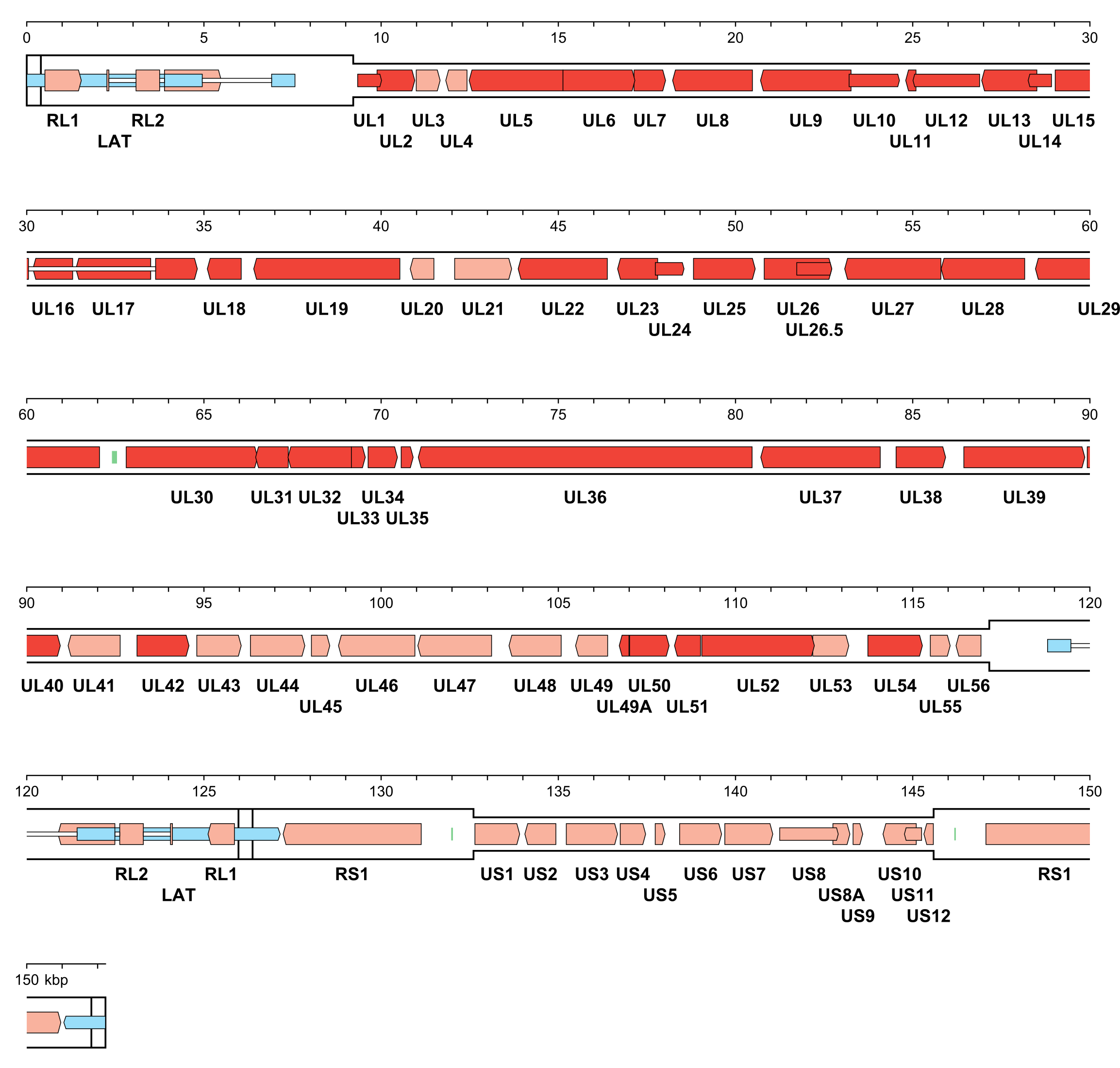
The number of orthoherpesvirus genes that encode functional proteins ranges from about 70 (e.g. VZV) to 170 (e.g. HCMV). The arrangement of protein-coding regions in a representative of each subfamily is shown in Figure 3.Orthoherpesviridae, Figure 4.Orthoherpesviridae and Figure 5.Orthoherpesviridae. Splicing occurs in a minority of genes, the proportion being lowest in members of the subfamily Alphaherpesvirinae. Ribosome profiling has identified a large number of additional open reading frames (ORFs) that may encode proteins in some family members, some of which have been detected by proteomics (Stern-Ginossar et al., 2012, Kronstad et al., 2013, Arias et al., 2014, Bencun et al., 2018, Whisnant et al., 2020). Except where these additional ORFs correspond to shorter or longer versions of established protein-coding regions, most are generally not conserved among related viruses and whether they encode functional proteins has not been determined. In addition to proteins, family members also encode RNAs that do not encode proteins, including long noncoding RNAs and micro RNAs (Stern-Ginossar et al., 2012, Cullen 2011, Gatherer et al., 2011, Rossetto et al., 2013, Ingolia et al., 2014, Wyler et al., 2017). The advent of high-throughput technology has enabled the sequencing of large numbers of strains of the most extensively studied viruses, including viral genomes in clinical material, and has led to a deeper understanding of the diversity and evolution of the family (Renner and Szpara 2018).
|
| Figure 3.Orthoherpesviridae. Genetic organisation of herpes simplex virus 1 strain 17 (JN555585, subfamily Alphaherpesvirinae). The inverted repeats (TRL, IRL, IRS and TRS, including a and a’) are shown in a thicker format than the unique regions (UL and US) (see Figure 2.Orthoherpesviridae for the relative arrangements of these regions in this class 4 genome). Functional protein-coding regions are indicated by open, coloured arrows, with bright red indicating genes shared with members of the other subfamilies and light red indicating other genes; gene nomenclature is shown below. Some protein-coding regions are designated by narrower coloured arrows merely to make their locations clearer. A noncoding RNA (the latency-associated transcript, LAT) mapping in two locations (one of which includes sequences near both genome termini) is indicated as narrower, blue-shaded arrows. Introns connecting protein-coding regions or LAT exons are shown as narrow white bars. Three origins of DNA replication are shown as green bars. |
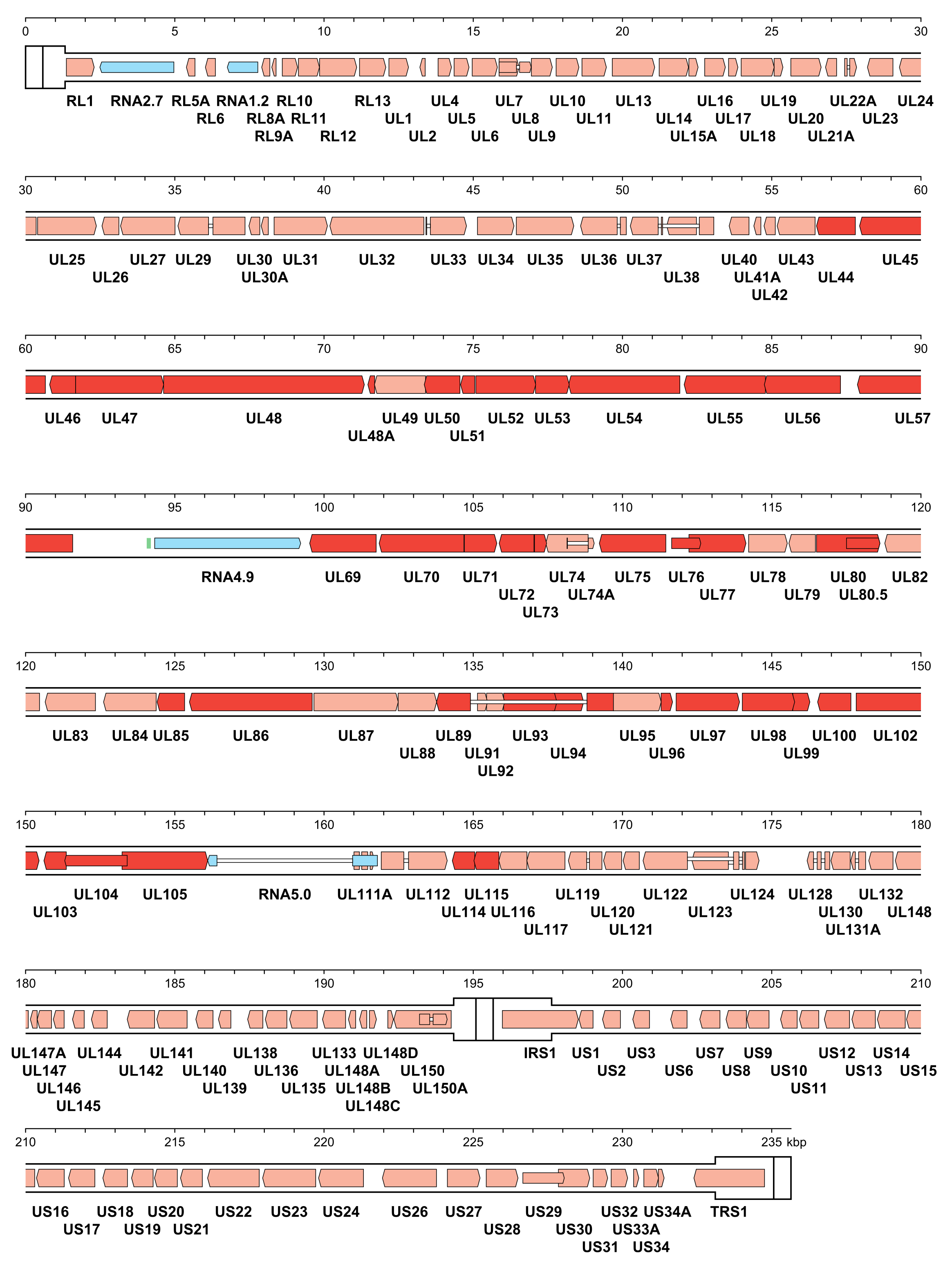
|
| Figure 4.Orthoherpesviridae. Genetic organisation of human cytomegalovirus strain Merlin (AY446894, subfamily Betaherpesvirinae). The inverted repeats (TRL, IRL, IRS and TRS, including a and a’) are shown in a thicker format than the unique regions (UL and US) (see Figure 2.Orthoherpesviridae for the relative arrangements of these regions in this class 4 genome). Functional protein-coding regions are indicated by open, coloured arrows, with bright red indicating genes shared with members of the other subfamilies and light red indicating other genes; gene nomenclature is shown below. Some protein-coding regions are designated by narrower coloured arrows merely to make their locations clearer. Noncoding RNAs (RNA2.7, RNA1.2, RNA4.9 and RNA5.0) are indicated as narrower, blue-shaded arrows. Introns connecting protein-coding regions or RNA5.0 exons are shown as narrow white bars. An origin of DNA replication is shown as a green bar. |
 |
| Figure 5.Orthoherpesviridae. Genetic organisation of Kaposi's sarcoma-associated herpesvirus strain GK18 (AF148805, subfamily Gammaherpesvirinae). The genome is depicted as the unique region (U) followed by a single copy of the terminal direct repeat (TR) in a thicker format. In genomic DNA, U is flanked on each side by variable numbers of TR, with the number of copies totalling 35–45, giving a genome length of approximately 170 kbp in this class 2 genome. Functional protein-coding regions are indicated by open, coloured arrows, with bright red indicating genes shared with members of the other subfamilies and light red indicating other genes; gene nomenclature is shown below. Some protein-coding regions are designated by narrower coloured arrows merely to make their locations clearer. Noncoding RNAs (T1.1 and T0.7) are indicated as narrower, blue-shaded arrows. Introns connecting protein-coding regions are shown as narrow white bars. Inverted repeats that may be associated with two origins of DNA replication are shown as green bars. |
A subset of 43 genes is detectably conserved among family members, although one or two have been lost in some lineages (Table 2.Orthoherpesviridae). These genes were presumably inherited from an ancestral herpesvirus and occur in six blocks that are arranged differently in members of the three subfamilies (Figure 6.Orthoherpesviridae) (McGeoch et al., 2006, Krug and Pellett 2021, Gompels et al., 1995, Davison 2010). Additional conserved genes may have been lost from members of the subfamily Alphaherpesvirinae but retained in members of the other subfamilies, following the early separation of this lineage (McGeoch et al., 1995). The conserved genes typically encode capsid proteins, components of the DNA replication and packaging machinery, nucleotide modifying enzymes, membrane proteins and tegument proteins, and, to a lesser extent, regulatory proteins. This reinforces the view that, despite their genetic diversity, family members share common features in many aspects of their replication strategies.
Table 2.Orthoherpesviridae. Shared genes in members of the family Orthoherpesviridae
Alphaherpes- virinae | Betaherpes- virinae | Gammaherpes- virinae | Protein name |
| herpes simplex virus 1* | human cytomegalovirus | Kaposi’s sarcoma-associated herpesvirus | |
| DNA replication machinery | |||
| UL30 | UL54 | ORF9 | DNA polymerase catalytic subunit |
| UL42 | UL44 | ORF59 | DNA polymerase processivity subunit |
| UL9 | lost | lost | DNA replication origin-binding helicase; present in members of the subfamily Alphaherpesvirinae and some members of the subfamily Betaherpesvirinae |
| UL5 | UL105 | ORF44 | Helicase-primase helicase subunit |
| UL8 | UL102 | ORF40 | Helicase-primase subunit |
| UL52 | UL70 | ORF56 | Helicase-primase primase subunit |
| UL29 | UL57 | ORF6 | Single-stranded DNA-binding protein |
| Enzymes peripheral to DNA replication | |||
| UL23 | lost | ORF21 | Thymidine kinase; present in members of the subfamilies Alphaherpesvirinae and Gammaherpesvirinae |
| UL39 | UL45 | ORF61 | Ribonucleotide reductase subunit 1; not an active enzyme in members of the subfamily Betaherpesvirinae |
| UL40 | lost | ORF60 | Ribonucleotide reductase subunit 2; present in members of the subfamilies Alphaherpesvirinae and Gammaherpesvirinae |
| UL50 | UL72 | ORF54 | Deoxyuridine triphosphatase; not an active enzyme in members of the subfamily Betaherpesvirinae |
| UL2 | UL114 | ORF46 | Uracil-DNA glycosylase |
| Processing and packaging of DNA | |||
| UL12 | UL98 | ORF37 | Deoxyribonuclease |
| UL15 | UL89 | ORF29 | DNA packaging terminase subunit 1 |
| UL28 | UL56 | ORF7 | DNA packaging terminase subunit 2 |
| UL6 | UL104 | ORF43 | Capsid portal protein |
| UL25 | UL77 | ORF19 | DNA packaging tegument protein UL25 |
| UL32 | UL52 | ORF68 | DNA packaging protein UL32 |
| UL33 | UL51 | ORF67A | DNA packaging protein UL33 |
| UL17 | UL93 | ORF32 | DNA packaging tegument protein UL17 |
| Egress of capsids from nucleus | |||
| UL31 | UL53 | ORF69 | Nuclear egress lamina protein |
| UL34 | UL50 | ORF67 | Nuclear egress membrane protein |
| Capsid assembly and structure | |||
| UL19 | UL86 | ORF25 | Major capsid protein |
| UL18 | UL85 | ORF26 | Capsid triplex subunit 2 |
| UL38 | UL46 | ORF62 | Capsid triplex subunit 1 |
| UL35 | UL48A | ORF65 | Small capsid protein |
| UL26 | UL80 | ORF17 | Capsid maturation protease |
| UL26.5 | UL80.5 | ORF17.5 | Capsid scaffold protein |
| Tegument | |||
| UL7 | UL103 | ORF42 | Tegument protein UL7 |
| UL11 | UL99 | ORF38 | Myristylated tegument protein |
| UL14 | UL96 | ORF35 | Tegument protein UL14 |
| UL16 | UL94 | ORF33 | Tegument protein UL16 |
| UL36 | UL48 | ORF64 | Large tegument protein |
| UL37 | UL47 | ORF63 | Tegument protein UL37 |
| UL51 | UL71 | ORF55 | Tegument protein UL51 |
| Surface and envelope | |||
| UL27 | UL55 | ORF8 | Envelope glycoprotein B |
| UL1 | UL115 | ORF47 | Envelope glycoprotein L |
| UL22 | UL75 | ORF22 | Envelope glycoprotein H |
| UL10 | UL100 | ORF39 | Envelope glycoprotein M |
| UL49A | UL73 | ORF53 | Envelope glycoprotein N |
| Control and modulation | |||
| UL13 | UL97 | ORF36 | Tegument serine/threonine protein kinase |
| UL54 | UL69 | ORF57 | Multifunctional expression regulator |
| Unknown | |||
| UL24 | UL76 | ORF20 | Nuclear protein UL24 |
In the HSV1 column, but not the HCMV and KHSV columns, genes essential for growth in vitro are shown in bold font.
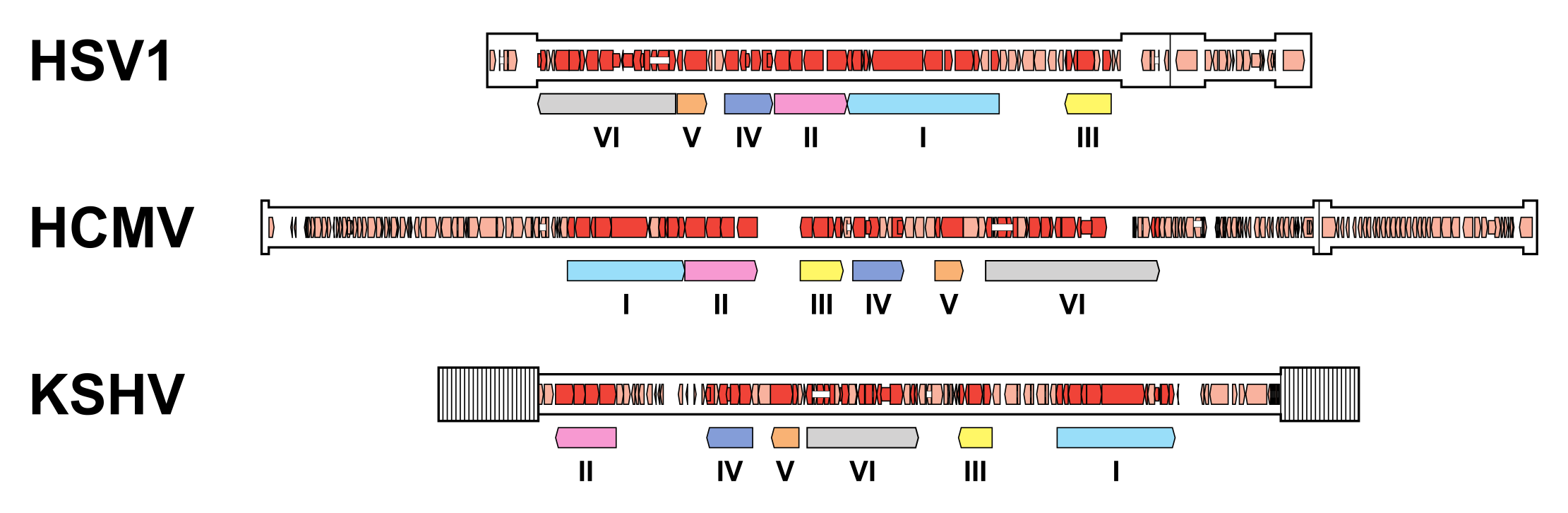 |
| Figure 6.Orthoherpesviridae. Organisation of conserved gene blocks in human herpesviruses representing the three subfamilies. The genomes illustrated are those of herpes simplex virus 1 strain 17 (HSV1; JN555585, subfamily Alphaherpesvirinae), human cytomegalovirus strain Merlin (HCMV; AY446894, subfamily Betaherpesvirinae) and Kaposi's sarcoma-associated herpesvirus strain GK18 (KSHV; AF148805, subfamily Gammaherpesvirinae). The conserved gene blocks (I-VI) are indicated by open, coloured arrows below the genomes, and are ordered relative to their arrangement in HCMV. Direct or inverted repeats are shown in a thicker format than unique regions. Functional protein-coding regions are indicated by open, coloured arrows, with bright red indicating genes shared with members of the other subfamilies and light red indicating other genes. Some protein-coding regions are designated by narrower coloured arrows merely to make their locations clearer. Introns connecting protein-coding regions are shown as narrow white bars. |
The precise details of the replication strategy during lytic infection vary from virus to virus, particularly in regard to the subfamily to which the virus belongs and the host cell type infected, but there are many general similarities (Figure 7.Orthoherpesviridae). The following brief description is based on well-studied family members, including HSV1, VZV and HCMV, and has been reviewed extensively (Damania and Cesarman 2013, Krug and Pellett 2021, Mettenleiter et al., 2009, Mocarski et al., 2013, Roizman et al., 2013, Yamanishi et al., 2013, Zerboni et al., 2014).
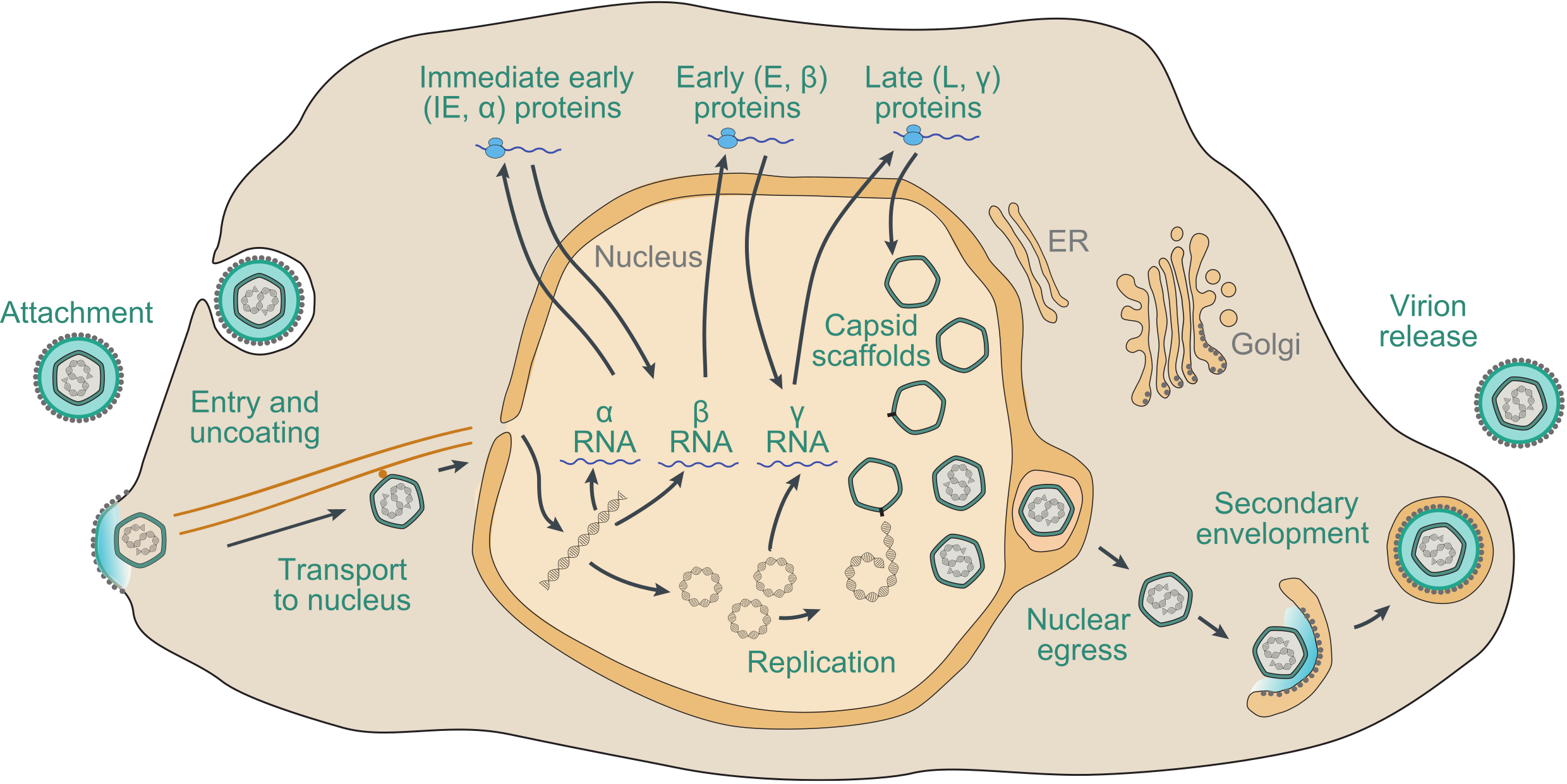 |
| Figure 7.Orthoherpesviridae. Schematic representation of the lytic replication cycle of a representative orthoherpesvirus in permissive cells, based on knowledge of herpes simplex virus 1, varicella-zoster virus and human cytomegalovirus. Further details are provided in this section. |
Adsorption of virions to the cell and penetration into the cytoplasm involve the interaction of multiple virion envelope proteins with multiple cell surface receptors. Entry takes place by membrane fusion either at the cell surface or following endocytosis of the attached virion, promoted by core glycoprotein complexes that include conserved glycoprotein B. The capsid is transported to the region of a nuclear pore by retrograde microtubule transport. Tegument proteins, many of which are of unknown function, are thought to modify cellular metabolism to favour viral replication. In permissive cells, entry of the genome into the nucleus is followed by a transcriptional cascade (Roizman and Campadelli-Fiume 2007). Immediate early (IE or α) genes, which are largely distinct among members of different subfamilies, regulate subsequent gene expression by transcriptional and post-transcriptional mechanisms. Early (E or β) genes encode the DNA replication complex and a variety of enzymes and other proteins involved in modifying host cell metabolism, and late (L or γ) genes primarily encode virion proteins. IE genes can be transcribed in the absence of de novo protein synthesis, E gene transcription is dependent on expression of IE proteins, and L gene transcription is dependent on viral DNA synthesis. With the exception of a few small, non-translated RNAs expressed by some members of the subfamily Gammaherpesvirinae, transcription involves host RNA polymerase II.
Viral DNA synthesis occurs from one or more origins of DNA replication, probably by a rolling-circle mechanism from circularised genomes, to generate concatemers. For HSV1, DNA replication requires seven viral proteins: an origin-binding helicase (encoded by gene UL9), a single-stranded DNA-binding protein (encoded by gene UL29), a DNA polymerase composed of a catalytic subunit (encoded by gene UL30) and a processivity subunit (encoded by gene UL42), and a helicase-primase complex comprising a primase subunit (encoded by gene UL52), a helicase subunit (encoded by gene UL5) and a third subunit (encoded by gene UL8) (Weller and Coen 2012). Orthologs of six of these genes are present in members of all three subfamilies, but UL9 has been lost in members of the subfamily Gammaherpesvirinae and in members of some lineages of the subfamily Betaherpesvirinae (Table 2.Orthoherpesviridae). Newly synthesized viral DNA is cleaved into unit-length genomes from concatemers and packaged into preformed immature capsids within the nucleus by processes that involve several viral proteins. The subsequent steps in virion morphogenesis are complex (Mettenleiter et al., 2009). Capsids bud through the inner nuclear membrane into the perinuclear space in a process termed primary envelopment, and are then de-enveloped by fusion with the outer nuclear membrane and released into the cytoplasm. Assembly of tegument proteins and secondary envelopment to generate mature virions appears to involve a Golgi or post-Golgi compartment.
Many viral genes are individually not required to achieve this basic lytic replication cycle in cell culture. Almost half the genes in HSV1, and an even higher proportion in viruses with larger genomes, such as HCMV, are in this category (Manservigi et al., 2010, Dunn et al., 2003, Yu et al., 2003). However, these genes have diverse and often significant functions in viral spread, host cell range and pathogenesis in vivo.
The alternative to lytic infection and consequent cell death is latent infection, whereby the virus enters a dormant state within the host with occasional reactivation leading to limited production of virions. Unlike lytic infection, the molecular mechanisms involved in latent infection appear to differ among members of different subfamilies and perhaps even among members of different genera within a subfamily. However, the cell lineages targeted for latency largely follow subfamily lines, with neuronal, myeloid and lymphoid cells most relevant for members of the subfamilies Alphaherpesvirinae, Betaherpesvirinae and Gammaherpesvirinae, respectively. Intensive studies have been carried out on HSV1 (Nicoll et al., 2012, Sawtell and Thompson 2021), HCMV (Dupont and Reeves 2016, Collins-McMillen et al., 2018) and EBV (Kang and Kieff 2015, Kempkes and Robertson 2015), but the means by which the latent state is established, maintained and reactivated in these viruses remains only partially understood. For HSV1, the latent state involves maintenance of the input genome as a circular episomal element in a chromatin-like state (Knipe and Cliffe 2008, Sawtell and Thompson 2016). Some latently infected neurons contain large numbers of HSV1 genome copies, suggesting that a latent state can be established after initiation of the productive cycle (Sawtell and Thompson 2004, Sawtell et al., 1998).
Like other large eukaryotic DNA viruses, some family members are in development as vectors for gene therapy (Argnani et al., 2005, Glorioso and Fink 2009), and some, particularly members of the subfamily Betaherpesvirinae, are being considered as platforms for self-disseminating vaccines against other pathogens (Murphy et al., 2016).
Biology
The range of vertebrate hosts in which family members have been discovered is very broad, extending from reptiles to birds and mammals, and the most extensively studied hosts harbour several different orthoherpesviruses (for example, humans are host to nine). In general, the natural host range of individual viruses is restricted, with most having evolved in association with their host species. As a result, orthoherpesviruses are highly adapted to their hosts, and severe infection is usually observed only in the foetus, the very young, the immunocompromised or following infection of an alternative host. Despite this general picture of coevolution, which in some instances may track host speciation, there is phylogenetic evidence that cross-species transmission has played an important part in the evolution of the family (McGeoch et al., 2000, Ehlers et al., 2008). Contemporary cross-species transmission has also been noted in some instances and is more likely among related hosts (e.g. among primates, equids or psittacine birds). These events may result in severe disease and death, and in some instances lead to misidentification of the natural host species (Tischer and Osterrieder 2010). Extinction has also played a major part in the shaping of the family, since most host species that have existed are extinct and will have taken their orthoherpesviruses with them. Moreover, it is possible for a virus to become extinct even when the host does not; this may be the reason why members of the subfamily Alphaherpesvirinae have not been found in murids and why members of the subfamily Betaherpesvirinae have not been discovered in equids or bovids. Given that the current level of sampling of family members in different host species is very low (probably representing much less than 1% of the number of orthoherpesvirus species in existence), the relative contributions of the various modes of evolution to the development of the family remain to be determined.
Host range varies considerably in experimental animal systems: some members of the subfamily Alphaherpesvirinae can infect a wide variety of animal species, whereas members of the subfamilies Betaherpesvirinae and Gammaherpesvirinae exhibit a very restricted experimental host range. Host range in cell culture also varies, though the same general rule holds true: members of the subfamily Alphaherpesvirinae can infect a variety of cells of diverse species, whereas members of the subfamilies Betaherpesvirinae and Gammaherpesvirinae exhibit greater restriction. The basis of host restriction is not well understood either in vivo or in cell culture. The presence and location of specific cell surface receptors determines tropism, and various other factors, including the interplay between innate cellular defences and immune evasion mechanisms, and the presence of host-specific transcription factors, determine the overall permissiveness of cells in vivo to productive infection (Adler et al., 2017).
Natural transmission routes range from aerosol spread (VZV, equine rhinopneumonitis virus in the species Varicellovirus equidalpha4; and feline viral rhinotracheitis virus in the species Varicellovirus felidalpha1) to intimate oral (EBV) or sexual contact (herpes simplex virus "type" 2 [HSV2] in the species Simplexvirus humanalpha2; and equine coital exanthema virus in the species Varicellovirus equidalpha3). Marek’s disease virus (MDV in the species Mardivirus gallidalpha2), gallid herpesvirus 3 (GaHV3 in the species Mardivirus gallidalpha3) and turkey herpesvirus (HVT in the species Mardivirus meleagridalpha1) are shed predominantly from the base of chicken feather follicles and transmitted as an airborne, inhaled dander. In addition to an exogenous route of infection, the HHV6A and HHV6B genomes are transmitted in about 1% of people via the host germ line when integrated into chromosomal telomeres (Clark 2016) via human telomere-like repeats near the genome termini (Gompels et al., 1995). Some family members, such as EHV1, can retain infectivity after long periods in drinking water (Dayaram et al., 2017), and this can be used as a method of delivering modified live vaccines, although the role of water in natural transmission is not clear (Coppo et al., 2012). Transmission can occur through direct mucosal contact or via droplets and fomites. Most family members establish a systemic infection via a cell-associated viraemia during primary infection. Infection with some members of the subfamily Alphaherpesvirinae (e.g. HSV1 and HSV2) may only produce localised lesions on the skin or mucosa of the oral or genital tracts, whereas more generalised infection can occur in young, immunocompromised or non-definitive hosts (James et al., 2014). A variety of immune evasion mechanisms have been identified, including those operating against complement, antibody, major histocompatibility complex class I presentation, natural killer cells and innate immune pathways (Arvin et al., 2007).
The ability of family members to establish life-long latent infection is considered to be key to their survival. Certain cell types that harbour latent virus have been identified, and this has suggested a general pattern in which most members of the subfamily Alphaherpesvirinae establish latent infection in neurons, members of the subfamily Betaherpesvirinae in haematopoietic cells, and members of the subfamily Gammaherpesvirinae in mononuclear cells, predominantly lymphocytes. However, this picture is based on a few selected examples, and there are reports of latent infection at other sites, such as members of the genus Mardivirus in the subfamily Alphaherpesvirinae establishing latency in lymphocytes (Calnek et al., 1981).
Antigenicity
Infected hosts produce antibodies and cell-mediated immune responses against many structural and non-structural viral proteins, and individual proteins harbour multiple epitopes against which antibody and cell-mediated responses may be elicited. Some envelope glycoproteins are targets for neutralizing antibodies, and a degree of cross-neutralization may be observed between related viruses within a genus (Arvin et al., 2007, MacLachlan et al., 2016). Cell-mediated immune responses, rather than neutralising antibodies, provide a stronger level of protection from recurrences and reinfection (Coppo et al., 2013, Kydd et al., 2003, Truong et al., 2019).
Derivation of names
Alphaherpesvirinae: from α (alpha), the first letter of the Greek alphabet.
Betaherpesvirinae: from β (beta), the second letter of the Greek alphabet.
Bossavirus: from the name of Dr Gregory D Bossart (1951 - 2019), former senior vice president and chief veterinary officer at Georgia Aquarium and expert in marine mammals (particularly dolphins) and their diseases. This genus contains a dolphin virus.
Cytomegalovirus: from the Greek κύτος (kútos), meaning a hollow, from which is derived cyto as in a cell, and μÎγα (méga), meaning large.
Gammaherpesvirinae: from γ (gamma), the third letter of the Greek alphabet.
Iltovirus: from infectious laryngotracheitis, the disease caused by infectious laryngotracheitis virus (ILTV in the species Iltovirus gallidalpha1), a member of the genus.
Lymphocryptovirus: from the Latin lympha, meaning water, and the Greek κρυπτÏŒς (kruptós), meaning concealed or cryptic.
Macavirus: from malignant catarrhal fever, the disease caused by the malignant catarrhal fever viruses (in the species Macavirus alcelaphinegamma1 and Macavirus alcelaphinegamma2), members of the genus.
Manticavirus: from the Latin mantica, meaning a small travelling bag, in reference to the marsupial pouch. This genus contains two marsupial viruses.
Mardivirus: from Marek’s disease (from the name of Dr József Marek (1868 - 1952)), which is caused by MDV, a member of the genus.
Muromegalovirus: from the Latin mus, meaning mouse, and the Greek μÎγα (méga) meaning large.
Orthoherpesviridae: from the Greek ὀρθός (ortho) meaning straight, right or proper, and ἕρπω, (hérpō) meaning to creep, in reference to the spreading blisters caused by HSV1 and HSV2.
Patagivirus: from the Latin patagium, meaning the gold edging or border on a woman’s tunic, in reference to the skin forming the surface of the wing in bats. This genus contains a bat virus.
Percavirus: from perissodactyls and carnivores, which are hosts of members of the genus.
Proboscivirus: from the Greek προβοσκίς (proboskís) and Latin proboscis meaning elephant’s trunk. This genus contains elephant viruses.
Rhadinovirus: from the Greek ῥαδινός (radinos), meaning slender or taper, in reference to the fragility of viral genomes during isolation.
Roseolovirus: from roseola, the disease caused by HHV6A, HHV6B and HHV7.
Quwivirus: from the Quechua quwi meaning guinea pig. This genus contains a guinea pig virus.
Scutavirus: from the Latin scutum, meaning shield, in reference to the shell of turtles and tortoises.
Simplexvirus: from the Latin simplex, meaning simple, presumably in reference to the appearance of the blisters caused by HSV1 and HSV2.
Varicellovirus: from the Latin varius, meaning diverse, and its incorporation into variola, meaning the pocks and pustules associated with smallpox, referring to the chickenpox rash typical of primary VZV infection.
Prior to the adoption by the ICTV of a binomial scheme, species names in the family Orthoherpesviridae consisted of three elements. The first was derived from the name of a taxon of the host that in its natural setting harbours the virus. The default taxon employed was that of family, and, except for herpesvirus species from humans, it ended in ‘-id’. Other exceptions were viral species from the family Bovidae, which were designated by host subfamily or genus, and from nonhuman primates, which were designated by host genus; these names ended in ‘-ine’. The second element was the word alphaherpesvirus, betaherpesvirus or gammaherpesvirus, depending on the subfamily to which the virus belongs. The third element was a numeral, or, in two cases, a numeral followed by a letter. The numeral was intended solely to provide a unique identifier, rather than to imply the existence of a complete or continuous series, or any particular relationship between viruses in different series that carry the same numeral. These numbers were chosen to avoid confusion in relation to the numerals used in virus names in the literature. Thus, ILTV was in the species Gallid alphaherpesvirus 1 and HHV6A was in the species Human betaherpesvirus 6A. This scheme was well-tolerated by herpesvirologists and served as a means for providing orthoherpesviruses with systematic virus names; thus, the systematic virus names for ILTV and HHV6A are gallid alphaherpesvirus 1 and human betaherpesvirus 6A.
The new species names consist of two words. The first is the name of the genus to which the species belongs, and the second consists of the old species name contracted by changing the initiating upper case character to lower case, deleting space characters, and deleting ‘herpesvirus’. Thus, the new names for Gallid alphaherpesvirus 1 and Human betaherpesvirus 6A are Iltovirus gallidalpha1 and Roseolovirus humanbeta6a, respectively. By this means, the new system for naming species in the family Orthoherpesviridae depends on the old system.
Subfamily demarcation criteria
The subfamilies were defined historically on the basis of broad biological features. They are now defined primarily on the basis of genetic content, which has proved generally, although not invariably, consistent with the original groupings. Thus, family members can be readily and reliably assigned to the subfamilies on the basis of: (i) comparison of the nucleotide or (more often) predicted amino acid sequences of conserved genes, and (ii) identification of particular genes or genetic properties that characterise a subfamily.
Genus demarcation criteria
The genera were defined historically on the basis of antigenic cross-reactivity and molecular criteria, primarily the size and structure of the genome. They are now defined primarily on the basis of genetic content.
Species demarcation criteria
Members of different orthoherpesvirus species have distinct epidemiological or biological characteristics and distinct genomes that represent independent replicating lineages. Replicating lineages of herpesviruses are identified primarily on the basis of information derived from genome sequences. Moreover, sequence information sufficient to demonstrate that a novel virus represents a replicating lineage distinct from members of known species is taken as evidence that the virus in question exists in nature and thus can be recognised as a member of an additional species. For some well-studied genes, there are levels of sequence difference beyond which the viruses in question are presumed to have distinct epidemiological and biological properties; such viruses can be reliably recognised as members of different species on the basis of limited sequence information. There are also closely related viruses that have relatively small differences in the sequences of individual genes, but these differences extend across the respective genomes in a manner indicating that they represent independent replicating lineages. These viruses also have distinct epidemiological and biological characteristics (e.g. host identity, pathogenic and epidemiological properties, and the absence or rarity of contemporary natural recombinants).
At present, sequence data are used flexibly to support taxonomic proposals on the basis of phylogenetic grouping of viruses. Each case is considered on its merits and does not require the availability of a complete genome sequence, does not depend on any specified gene or group of genes, and does not specify genetic distance thresholds for differentiating taxa.
Relationships within the family
The phylogeny and evolution of the family have been studied in depth by sequence analysis (McGeoch et al., 2006, Davison 2010, McGeoch et al., 1995, Brito and Pinney 2020). The three subfamilies are thought to have diverged over a timescale of a few hundred million years (McGeoch et al., 2006, McGeoch et al., 1995, McGeoch et al., 2000). The phylogenies illustrated in Figure 1.Alphaherpesvirinae, Figure 1.Betaherpesvirinae and Figure 1.Gammaherpesvirinae were generated using a concatenation of the amino acid sequences predicted for six conserved genes (Table 3.Orthoherpesviridae); details of the input data are provided in Resources 1–4. These phylogenies include only species for which the sequences of these six genes are available (typically in the context of a complete genome) and exclude species that have been classified on the basis of other criteria (usually a subset of these genes or alternative genes, or criteria that did not involve sequence data).
Table 3.Orthoherpesviridae. Conserved genes used for phylogenetic analysis
| Protein name | Alphaherpesvirinae | Betaherpesvirinae | Gammaherpesvirinae |
| Virus | herpes simplex virus 1 | human cytomegalovirus | Kaposi's sarcoma-associated herpesvirus |
| Uracil-DNA glycosylase | UL2 | UL114 | ORF46 |
| Helicase-primase helicase subunit | UL5 | UL105 | OR44 |
| DNA packaging terminase subunit 1 | UL15 | UL89 | ORF29 |
| Major capsid protein | UL19 | UL86 | ORF25 |
| Envelope glycoprotein B | UL27 | UL55 | ORF8 |
| DNA polymerase catalytic subunit | UL30 | UL54 | ORF9 |
The gene names for one member of each subfamily are listed.
Relationships with other taxa
The family Orthoherpesviridae is placed with two other families in the order Herpesvirales on the basis of a characteristic virion morphology: Alloherpesviridae (members of which infect fish and amphibian hosts) and Malacoherpesviridae (members of which infect invertebrate hosts). Common demarcation criteria apply across the order.
The evolutionary connections of members of the family to members of the two other families in the order Herpesvirales are reflected not only in virion structure but also in conservation of DNA packaging terminase subunit 1, the ATPase-bearing component of the DNA packaging terminase complex responsible for packaging viral DNA into nascent capsids (Davison 1992) (Table 2.Orthoherpesviridae). Similarities are also evident in a number of small motifs in other proteins (Mushegian et al., 2018). The diversity of the viruses in the order Herpesvirales has meant that criteria such as serology or nucleic acid hybridization have had limited value in determining relationships between different viruses, and the construction of a satisfactory taxonomic structure has been a significant challenge.
Very distant relationships between members of the order Herpesvirales and bacteriophages in the order Caudovirales have long been recognised from sequence, structural and functional similarities (Davison 1992, Cheng et al., 2004, Liu and Mushegian 2004, Baker et al., 2005, Rixon and Schmid 2014). The combination of these relationships strongly indicates that a part of the genetic inheritance of the two orders is shared (McGeoch et al., 2006). The recent extension of the number of available taxonomic ranks for viruses (International Committee on Taxonomy of Viruses Executive Committee 2020) has enabled this to be reflected in the classification, with the order Herpesvirales nested in the class Herviviricetes, phylum Pepliviricota, kingdom Heunggongvirae and realm Duplodnaviria. However, only the levels of order and kingdom contain more than a single taxon at present.
Members of the family Orthoherpesviridae possess several genes (e.g. encoding enzymes or immunomodulatory factors) that are related to cellular genes and are assumed to have been gained by capture or convergent evolution. Similar genes in other virus families probably indicate independent capture or convergent evolution, perhaps due to the effects of similar selective environments (e.g. replication in leukocytes), and it appears that such events have occurred in some circumstances even within the family Orthoherpesviridae. As a result, genes in this category are best avoided for the purposes of classification.
Related, unclassified viruses
Unclassified viruses for which partial, and often short, sequences are available are too numerous to be listed. Many of the unclassified viruses listed in the 9th Report are no longer available and lack associated sequence data, and therefore cannot be classified.