

Family: Redondoviridae
Arwa Abbas, Louis J. Taylor, Ronald G. Collman and Frederic D. Bushman
The citation for this ICTV Report chapter is the summary published as Abbas et al. (2021):
ICTV Virus Taxonomy Profile: Redondoviridae, Journal of General Virology, 102 (1): 001526
Corresponding author: Frederic D. Bushman ([email protected])
Edited by: Balázs Harrach and Stuart G. Siddell
Posted: November 2020
PDF: ICTV_Redondoviridae.pdf
Summary
Redondoviridae is a newly-identified family of viruses whose members have circular DNA genomes of about 3.0 kb (Table 1. Redondoviridae). The genome is inferred to be single stranded by analogy with other viruses of the circular, Rep-encoding single-stranded (CRESS) group. Redondoviruses were discovered through metagenomic sequencing methods. As a result, their definitive host is unknown, but they are inferred to replicate primarily in humans. Redondoviruses have been found predominantly in oro-respiratory specimens. Redondoviruses are not known to cause any diseases in humans, though elevated oro-respiratory redondovirus DNA levels are associated with periodontitis and in patients treated for critical illness.
Table 1. Redondoviridae. Characteristics of members of the family Redondoviridae
| Characteristic | Description |
| Typical member | human respiratory-associated brisavirus, isolate LC (KY052047), species Torbevirus brisa |
| Virion | Unknown |
| Genome | Circular, about 3.0 kb, inferred to be single stranded DNA |
| Replication | Unknown |
| Translation | Unknown |
| Host range | Presumed to be human-specific |
| Taxonomy | Realm Monodnaviria, kingdom Shotokuvirae, phylum Cressdnaviricota, class Arfiviricetes, order Recrevirales; the genus Torbevirus includes two species |
Virion
Morphology
The physical structure and properties of redondoviruses have not yet been documented. The capsid protein likely derives from the largest open reading frame (Cp), although the number and orientation of capsomer units is unknown.
Physicochemical and physical properties
None reported.
Nucleic acid
The genome is a circular DNA of about 3.0 kb, inferred to be single stranded by analogy to related viruses. The genome has 3 ORFs in opposite orientations. One ORF (Cp) encodes the putative capsid protein, another (Rep) encodes the Replication-associated protein, and a third ORF has no homology to any known protein, but is found in all redondovirus genomes. A stem-loop structure is found before the beginning of the Rep coding sequence (Table 1. Redondoviridae, Figure 1. Redondoviridae). The inverted repeat forming the stem is variable in length, while the nonanucleotide motif (5′-TATTATTAT-3′) forming the loop is largely conserved amongst identified genomes. Additionally, imperfect direct repeats of approximately 6 nucleotides are observed downstream of the stem loop (Abbas et al., 2019). This stem loop structure is a likely candidate for the origin of replication, as described for the related circoviruses (Mankertz et al., 1997).
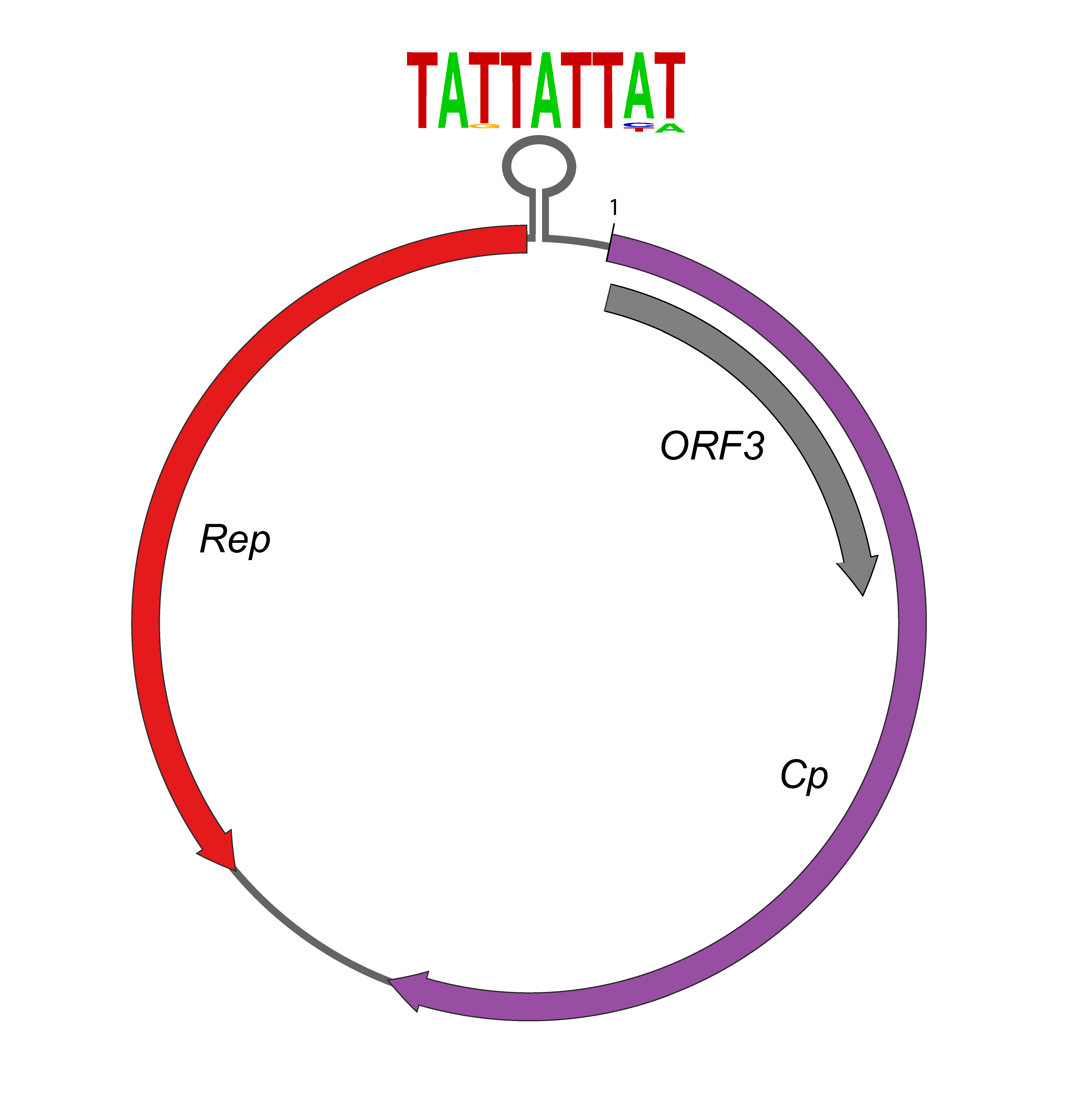 |
| Figure 1. Redondoviridae. Redondovirus genome structure. The brisavirus genome illustrated is 3.0 kb with 3 ORFs separated by two intergenic regions. The largest ORF (purple) encodes the putative capsid (Cp) protein, the second largest (red) encodes the Replication-associated (Rep) protein, and the third (grey) encodes a protein with no known sequence or structural homologue. A stem-loop structure is predicted to form before the beginning of the Rep coding sequence. The loop contains a conserved nonanucleotide motif and is thought to be the Rep recognition site and origin of viral replication. The height of the letters in the motif represents their proportion in different redondovirus genomes. |
Proteins
The capsid protein (Cp, Figure 2B. Redondoviridae) contains a basic amino-terminus. Protein modelling using PHYRE2 (Kelley et al., 2015) predicted a fold similar to coat proteins of plant-infecting, positive-sense RNA viruses in the family Tombusviridae.
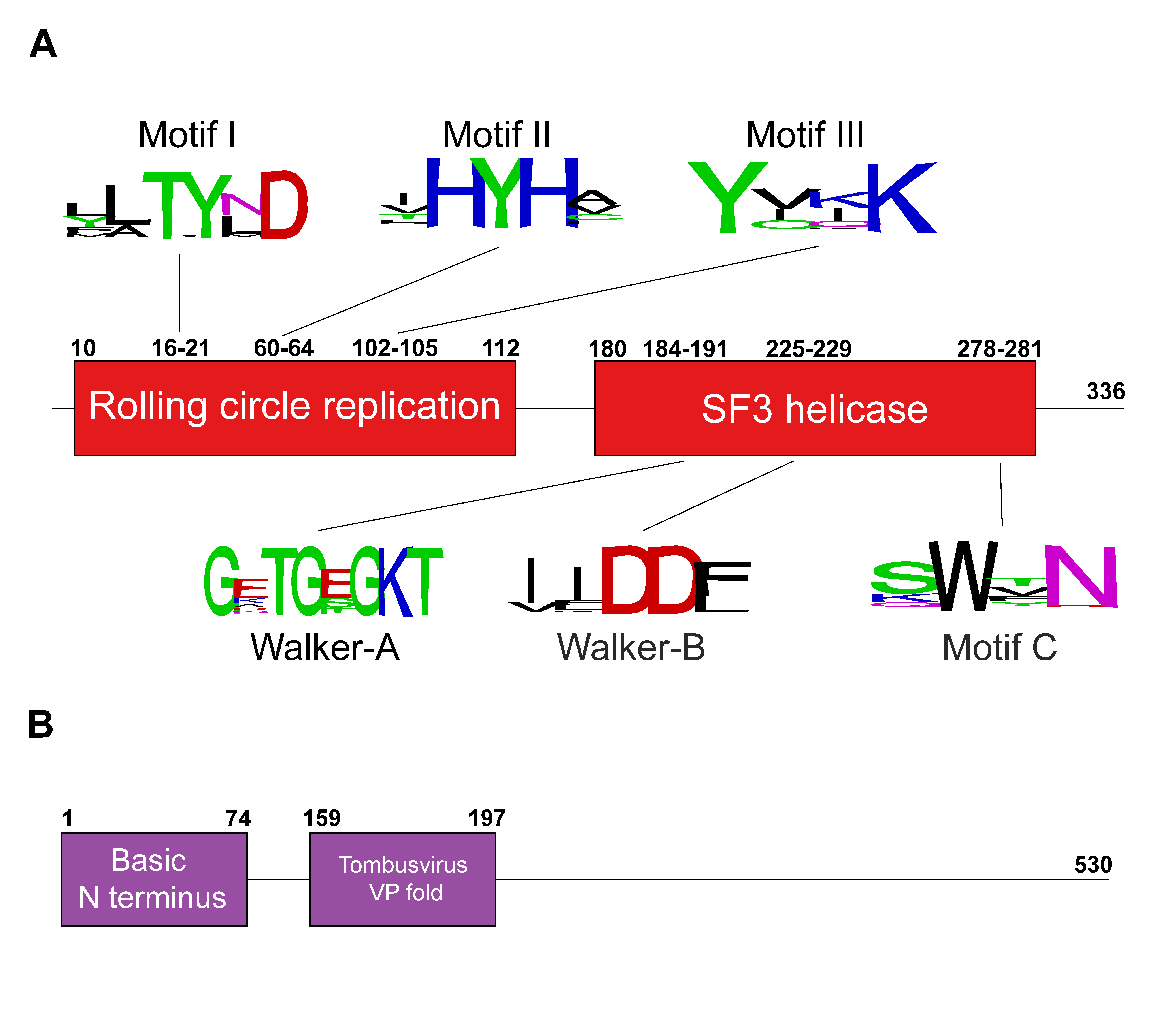 |
| Figure 2. Redondoviridae. Amino acid sequence motifs found in redondovirus proteins. A: Amino acid sequence motifs associated with rolling circle replication and superfamily 3 (SF3) helicase function. The amino-acid positions are indicated on the human lung-associated brisavirus AA (GenBank: MK059754) Rep protein sequence. The height of each letter represents its proportion amongst all the redondovirus genomes described thus far. Amino acids are coloured according to their hydrophobicity and charge. B: Domains found in the putative redondovirus capsid protein. Shown are a basic amino-terminal domain and a predicted tombusvirus coat protein-like (VP) fold. The positions are shown for human lung-associated brisavirus AA. |
Lipids
None reported.
Carbohydrates
None reported.
Genome organization and replication
Virus genomes encode a 334–363 amino acid replication associated protein (Rep) and a 449–531 amino acid capsid protein (Cp). All genomes also contain a third ORF (ORF3), which, if translated, would encode a 200 amino acid protein. Pairwise amino acid identities of Cp between different viruses are 67.5%–99.6% (median 82.3%) while Rep is less conserved at 36.6%–99.7% amino acid identity (median 54%) (Abbas et al., 2019).
The Rep protein (Figure 2A. Redondoviridae) contains two domains, the rolling-circle replication (RCR) endonuclease and the superfamily 3 helicase (SF3) domains. These domains, which are found in many ssDNA and RNA viruses, are used to determine phylogenetic relationships amongst viral taxa (Koonin et al., 2020). The N-terminal RCR domain is characterized by conserved motifs, known as Motif I, Motif II and Motif III. Motif I is thought to recognize the origin of replication, Motif II contains histidine residues required for coordination of divalent metal ions and Motif III contains the catalytic tyrosine residues important in Rep function (Ilyina and Koonin 1992, Rosario et al., 2012). The C-terminus of the Rep protein contains the SF3 helicase domain containing Walker-A and Walker-B motifs, of the NTP-binding pattern (Gorbalenya et al., 1990), as well as Motif C.
The replication cycle is presumed to follow that of other circular ssDNA viruses, such as circoviruses, via rolling-circle replication (RCR) (Faurez et al., 2009). In this model, upon entry of a host cell and uncoating of the virion, the viral ssDNA genome is converted to dsDNA by host polymerases, as the viral genome itself does not encode a DNA polymerase. The viral Rep protein binds in a sequence-specific manner to the stem-loop structure and nicks dsDNA, creating a free 3′-hydroxyl end from which viral DNA synthesis can begin. The Rep protein, meanwhile, remains covalently bonded to the 5′-phosphate end. After one round of genome synthesis, the Rep protein releases one ssDNA genome, and the dsDNA template is regenerated for additional rounds of RCR.
Biology
Redondovirus nucleic acid sequences have been detected in both healthy and diseased humans. The first reported genome was discovered in the respiratory tract of a febrile patient (Cui et al., 2017) who tested negative for a limited panel of other pathogens. Subsequently, full-length genomes were found in bronchoalveolar lavage from organ transplant donor lungs, lung transplant recipients and patients with sarcoidosis (Abbas et al., 2017, Abbas et al., 2019, Clarke et al., 2018). Redondoviruses were also detected in sputum samples from a patient with respiratory symptoms in the absence of any other identified respiratory pathogen (Lázaro-Perona et al., 2020). A single isolate has been reported from bees (Kraberger et al., 2019), but this may not be its definitive host since its sequence (MH973742) is 98.8% identical to one derived from a human sample (MK059754).
A large set of metagenomic datasets from humans, animals and the environment were analysed for redondoviral genome sequences. This screen surveyed 51 non-human organisms and environments, as well as sequencing reagent controls. Viral sequences were only detected with certainty in human samples, primarily from the oral cavity, nasopharynx and lung, and also infrequently in the gut (Abbas et al., 2019).
Molecular-based detection by qPCR revealed that redondovirus nucleic acid was present at relatively high levels in the upper and lower respiratory tract of hospitalized patients in medical intensive care units, compared to levels found in the upper respiratory tract of healthy adults. In patients providing serial samples, virus nucleic acid was detectable over a period of 2–3 weeks, suggesting persistent colonization or infection. One patient was positive when sampled over a two year interval. Overall, prevalence in oropharyngeal samples of healthy U.S. adults was reported to be about 15% (Abbas et al., 2019).
Analysis of sequences (Abbas et al., 2019) from two metagenomic studies of the oral cavity (Shi et al., 2015, Califf et al., 2017) showed that the presence and abundance of redondoviral genome sequence was associated with periodontitis.
For the associations of redondoviruses with periodontitis and critical illness, it is not known whether viral infection has a role in causing or maintaining the disease state.
In virome datasets from metagenomic samples, members of the family Redondoviridae significantly co-occurred with members of another ubiquitous ssDNA family, Anelloviridae (Abbas et al., 2019). However, it is undetermined whether redondoviruses require the presence of anelloviruses to replicate in the human host.
In summary, members of the family Redondoviridae are not proven to be the etiological agent of any disease, but show strong evidence of replicating in the human respiratory tract.
Derivation of names
Brisavirus: from the Spanish brisa for “breeze”, alluding to the prevalence of these viruses in the respiratory tract
Redondovirus: from the Spanish redondo for “round”, alluding to the circular nature of the DNA genome
Torbevirus: from the Spanish torbellino for “whirlwind”, as this incorporates both the circular nature of the virus genome and prevalence in the respiratory tract
Vientovirus: from the Spanish viento for “wind”, alluding to the prevalence of these viruses in the respiratory tract
Relationships within the family
The family includes a single genus with two species, Brisavirus and Vientovirus, members of which belong to distinct clades upon phylogenetic analysis of their Rep protein sequences (Figure 3. Redondoviridae).
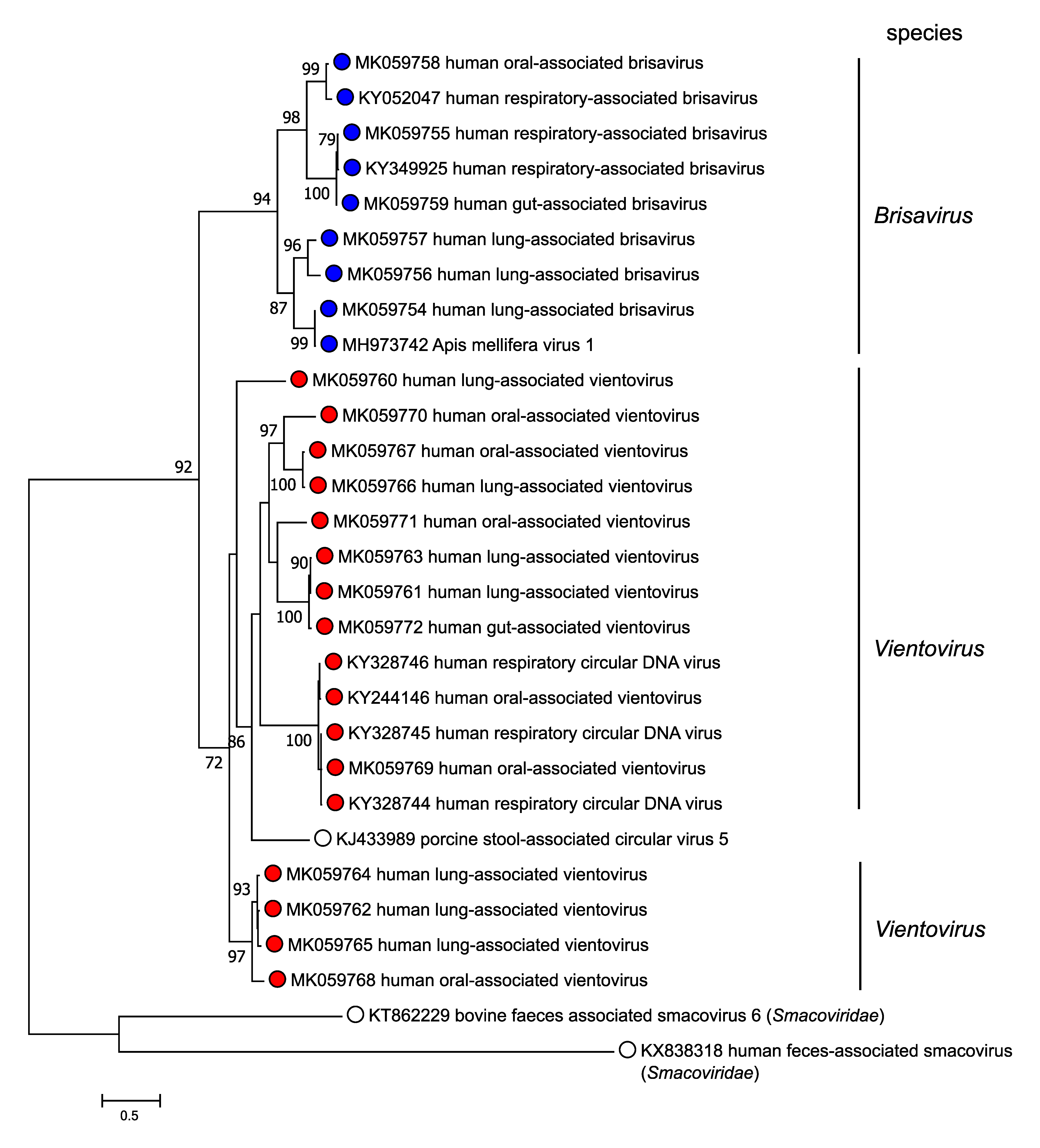 |
| Figure 3. Redondoviridae. Phylogeny of members of the family Redondoviridae. Rep proteins of porcine stool-associated circular virus 5, apis mellifera virus 1, eight brisaviruses and 17 vientoviruses were aligned with MUSCLE (Edgar 2004), trees built using PHYML with branch support determined by approximate likelihood ratio test (Guindon et al., 2010) and visualized using FigTree. Rep proteins from two members of the family Smacoviridae are included as an outgroup. This phylogenetic tree and corresponding sequence alignment are available to download from the Resources page. |
Redondovirus genomes share limited homology (<20% genome coverage) with an unclassified virus, porcine stool-associated circular virus 5 (PoSCV-5) (Cheung et al., 2014). In the case of PoSCV-5, while the Rep protein identity and genome size fall within the bounds of Redondoviridae family members (Figure 3. Redondoviridae), the capsid protein is much less similar (40–50% identity). In comparison, there is >70% identity among the capsids of members of the family Redondoviridae. PoSCV-5 also lacks the conserved ORF3. PoSCV-5 remains unclassified, although future genomes discovered may provide evidence for or against including PoSCV-5 in the family. Additionally, a full-length viral genome (MH973742, apis mellifera virus 1) isolated from a honey bee hemolymph sample (Kraberger et al., 2019) encodes Rep and capsid proteins with high sequence identity to those of human brisaviruses; the definitive host is unknown.
Relationships with other taxa
Assessment of replication-associated protein (Rep) phylogeny (Figure 4. Redondoviridae), reveals that members of the family Redondoviridae form a clade distinct from other taxa within the phylum Cressdnaviricota. Similar results are obtained for the capsid protein. Currently, Redondoviridae is the only family within the order Recrevirales (Koonin et al., 2020).
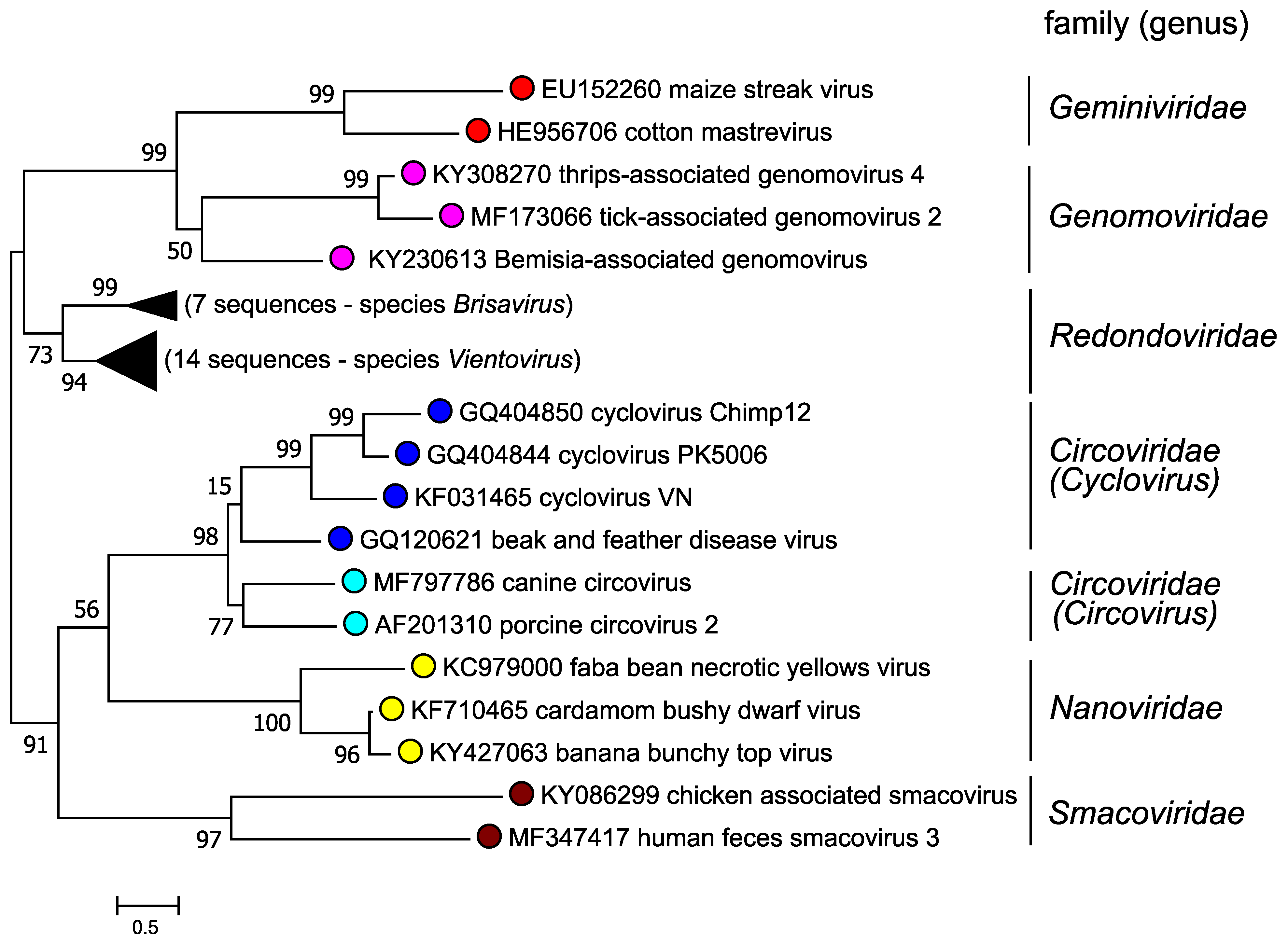 |
| Figure 4. Redondoviridae. Phylogeny of CRESS DNA viruses. Rep amino acid sequences of representatives of different CRESS virus families were aligned using MUSCLE (Edgar 2004). Trees were built using PHYML with branch support determined by the approximate likelihood ratio test (Guindon et al., 2010), and visualized using FigTree. Nodes are labelled with probabilities converted to percentages. Accession numbers for isolates in the family Redondoviridae were as in Figure 3. Redondoviridae. This phylogenetic tree and corresponding sequence alignment are available to download from the Resources page. |
Members of the family Redondoviridae share certain features with other families within the phylum Cressdnaviricota (Table 2. Redondoviridae).
Table 2. Redondoviridae. Comparison of genomic features between members of the family Redondoviridae and members of other CRESS DNA viral families
| Features | |||||
| Family | Size (kb) | ORFs | ORF orientation | Origin sequence | Origin location |
| Redondoviridae | 3.0–3.1 | Cp, Rep, ORF3 | Opposite | TATTATTAT | Noncoding (upstream)/ in Rep |
| Circoviridae | 1.7–2.0 | Cp, Rep, ORF3, ORF4 | Opposite | TAGTATTAC | Noncoding (upstream)/ in Rep |
| Nanoviridae | 1.0*6 segments | Cp, Rep, others | Segmented | TATTATTAC | Noncoding (upstream) |
| Geminiviridae | 2.5–3.0 | Cp, Rep, others | Opposite (or segmented) | TAATATTAC | Noncoding (upstream) |
| Genomoviridae | 2.1–2.2 | Cp, Rep | Opposite | TAATATTAT | Noncoding (upstream) |
| Smacoviridae | 2.6–2.9 | Cp, Rep | Opposite | NAGTATTAC | Noncoding (downstream) |
| Bacilladnaviridae | 5.5–6, portion of genome is dsDNA | Cp, Rep, 1–2 others | Opposite | Not reported | Not reported |
Related, unclassified viruses
| Virus name | Accession number | Virus abbreviation |
| porcine stool-associated circular virus 5 | KJ433989 | PoSCV-5 |