Vertebrate Prions
Chapter Version: ICTV Ninth Report; 2009 Taxonomy Release
Prions are proteinaceous infectious particles that lack nucleic acids. In mammals, prions are composed largely, if not entirely, of a pathogenic isoform of the host-encoded cellular prion protein (PrP). Neither prion-specific nucleic acids nor virus-like particles have been detected in highly purified, infectious preparations.
In fungi, evidence for nine different prions has been accumulated. Those studies are summarized in the section on Fungal Prions.
General description of the mammalian prions
The mammalian prions cause scrapie and other related neurodegenerative diseases of animals and humans (Table 1). The prion diseases are also referred to as the transmissible spongiform encephalopathies (TSEs). TSEs are inevitable fatal and no treatment is available for the afflicted species.
Prions are composed of an abnormal, pathogenic PrP isoform, denoted PrPSc. The “Sc” superscript was initially derived from the term scrapie, because scrapie was the prototypic prion disease. Since all of the known prion diseases (Table 1) of mammals involve aberrant metabolism of PrP similar to that observed in scrapie, the “Sc” superscript was suggested for all pathogenic PrP isoforms. In this context, the “Sc” superscript is used to designate the scrapie-like isoform of PrP; for those who desire a more general derivation “Sc” can equally well be derived from the term “prion sickness” (see Table 2).
A post-translational process converts the normal cellular isoform of the protein, designated PrPC, into PrPSc. Attempts to identify posttranslational modifications that distinguish PrPSc from PrPC have not been successful; moreover, PrPC and PrPSc are encoded by the same single copy chromosomal gene. The conformations of the two PrP isoforms are profoundly different; PrPC has little β-sheet while PrPSc has a high β-sheet content.
Like viruses, prions are infectious because they stimulate a process by which more of the pathogen is produced. As prions or viruses accumulate in an infected host, they eventually cause disease. Both prions and viruses exist in different varieties or subtypes that are called strains. But many features of prion structure and replication distinguish them from viruses and all other known infectious pathogens.
Prions differ from viruses and viroids since they lack a nucleic acid genome that directs the synthesis of their progeny. Prions are composed of an abnormal isoform of a cellular protein whereas most viral proteins are encoded by the viral genome and viroids are devoid of protein. Prions can exist in multiple molecular forms, whereas viruses exist in a single form with a distinct ultrastructural morphology. Prions are non-immunogenic, in contrast to viruses, which almost always provoke an immune response. An enlarging body of evidence argues that strains of prions are enciphered in the conformation of PrPSc; in contrast, strains of viruses and viroids have distinct nucleic acid sequences that produce pathogens with different properties.
Table 1 The prion diseases
| Disease (Abbreviation) | Natural host | Prion | Pathogenic PrP isoforms |
| Scrapie | sheep and goats | Scrapie prion | OvPrPSc |
| Transmissible mink encephalopathy (TME) | mink | TME prion | MkPrPSc |
| Chronic wasting disease (CWD) | mule deer and elk | CWD prion | MDePrPSc |
| Bovine spongiform encephalopathy (BSE) | cattle | BSE prion | BoPrPSc |
| Feline spongiform encephalopathy (FSE) | cats | FSE prion | FePrPSc |
| Exotic ungulate encephalopathy (EUE) | nyala, oryx and greater kudu | EUE prion | UngPrPSc |
| Kuru | humans | Kuru prion | HuPrPSc |
| Sporadic Creutzfeldt–Jakob disease (sCJD) | humans | sCJD prion | HuPrPSc |
| Familial Creutzfeldt–Jakob disease (fCJD) | humans | fCJD prion | HuPrPSc |
| Iatrogenic Creutzfeldt–Jakob disease (iCJD) | humans | iCJD prion | HuPrPSc |
| Variant Creutzfeldt–Jakob disease (vCJD) | humans | vCJD prion | HuPrPSc |
| Gerstmann–Sträussler Scheinker syndrome (GSS) | humans | GSS prion | HuPrPSc |
| Sporadic fatal familial insomnia (iFFI) | humans | sFFI prion | HuPrPSc |
| Familial fatal familial insomnia (fFFI) | humans | fFFI prion | HuPrPSc |
Table 2 Glossary of prion terminology
| Term | Description |
| Prion | A proteinaceous infectious particle that lacks nucleic acid. Prions are composed largely, if not entirely, of PrPSc molecules |
| PrPSc | Abnormal, pathogenic isoform of the prion protein that causes sickness. This protein is the only identifiable macromolecule in purified preparations of prions |
| PrPC | Cellular isoform of the prion protein |
| PrP27-30 | Digestion of PrPSc with proteinase K generates PrP27-30 by hydrolysis of the N-terminal part |
| PrPres | Alternative designation for PrPSc, that has been proposed to generalize the term for all types of TSEs and not only scrapie |
| PrPsen | Host-encoded PrPC that is sensitive to hydrolysis by Proteinase K |
| rPrP | Recombinant PrP generated in E. coli that lacks the two sugar moieties and the GPI anchor |
| PRNP | Human PrP gene located on chromosome 20 |
| Prnp | Mouse PrP gene located on syntenic chromosome 2. Prnp controls the length of the prion incubation time and is congruent with the incubation time genes Sinc and Prn-i |
| Prnp°/° | PrP-deficient mice that are resistant to prions |
| PrP amyloid | Fibril of PrP fragments derived from PrPSc by proteolysis. Plaques containing PrP amyloid are found in the brains of some mammals with prion disease |
| Prion rod | An amyloid polymer composed of PrP27-30 molecules. Created by detergent extraction and limited proteolysis of PrPSc |
| Protein X | A hypothetical macromolecule that is thought to act like a molecular chaperone in facilitating the conversion of PrPC into PrPSc |
Mammalian prion diseases
Scrapie was reported for the first time 250 years ago in Great Britain and it affects mainly sheep and goats. The most efficient proof for the transmissibility of the disease was demonstrated when more than 1,500 sheep came down after vaccination against looping-ill virus with a formalin-treated vaccine derived from ovine lymphoid tissue unknowingly infected with scrapie. More recently, there has also been a recrudescence of scrapie outbreaks among European sheep flocks. The mechanism on how the disease can be transmitted from one animal to another is still unclear. Chronic inflammation seems to alter the tropism of prion infectivity to organs hitherto believed prion-free, like mammary glands. This raised concerns about scrapie transmission through milk.
The commonest prion disease is the bovine spongiform encephalopathy, more commonly known as “mad cow disease”, which appeared for the first time in Great Britain in 1986. Since then more than 280,000 cattle have died from this disease. The source of the disease was attributed to animal-derived food supplements that were fed as a nutritional supplement to cattle. The disease has become quite rare since 2003 and only 37 cases were reported in the UK in 2008 (http://www.oie.int/).
Another animal prion disease that has evolved in the United States of America is Chronic Wasting Disease (CWD) in captive deer, with the first case observed in 1967 in a research facility in Fort Collins. Since 1980, cases involving free-ranging animals in Colorado and Wyoming have been reported and to date CWD has been observed in 14 states and two Canadian province. The known natural hosts of CWD are mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus), Rocky Mountain elk (Cervus elaphus nelsoni) and Shira’s moose (Alces alces Shirasi). The disease is believed to be horizontally transmitted between cervids with high efficiency.
Transmissible mink encephalopathy (TME), feline spongiform encephalopathy (FSE) and exotic ungulate encephalopathy (EUE) are all thought to occur after the consumption of prion-infected materials.
Human prion diseases
The human prion diseases comprise Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker (GSS) syndrome, Fatal Familial Insomnia (FFI) and Kuru. Epidemiologically, CJD is classified as sporadic (sCJD), familial (fCJD), iatrogenic (iCJD) and variant (vCJD). Even the most frequent form, sCJD, is very rare, with an incidence of 0.4–1.8 person in one million people each year worldwide. Typically, humans with an age between 55 and 70 years are affected. The source of the disease is still unknown, but might have its origin in a spontaneous conversion of PrPC into PrPSc as a rare stochastic event or in somatic mutations in the PRNP gene that encodes the prion protein.
Iatrogenic CJD cases are rare. These cases originate from inoculation of prions through human pituitary-derived growth hormone, cornea transplants, duramater grafts, or cerebral electrode implants. vCJD was recently identified in humans and has affected ~200 victims worldwide since 2006. The histopathology of this disease is reminiscent of that of BSE indicating that this disease has originated by the transmission of BSE to humans. Kuru occurred among the people of the Fore Tribe in Papua New Guinea and was horizontally transmitted between humans by cannibalistic rituals. Since this practice was stopped by the government no new cases have been reported in individuals born after that date.
Familial forms of CJD as well as all cases of GSS and FFI are caused by mutations in PRNP.
Nomenclature of the mammalian prions
The terminology for prions is still evolving (Table 2). While some terms are borrowed from infectious diseases caused by viruses, others are taken from genetics and still others from the biology of protein structure as well as neuropathology. This new area of biological investigation, which has such diverse roots, creates some unique problems with terminology.
Prion properties
Physicochemical and physical properties
The mature human prion protein consists of 208 amino acids. The N-terminal signal sequence that targets the protein into the endoplasmic reticulum is cleaved off in the mature form and a C-terminal polypeptide of 23 amino acids is replaced by the addition of a glycosylphospatidylinositol anchor (GPI) to the C-terminal serine residue. The protein also contains a single disulfide bridge which is formed between residues Cys-179 and Cys-214 and two N-linked glycosylation sites at positions Asn-181 and Asn-197 (Figure 1A).
The N-terminal part of the prion protein contains several Gly-Pro-rich tandem repeats, which vary in number, length and composition between species and are flanked by two positively charged clusters of amino acids 23–27 (CC1) and 95–110 (CC2). The center of the protein contains a hydrophobic stretch of amino acids 111–134 with the palindrome sequence AGAAAAGA. This region is highly conserved between species and is even present in fish and another member of the prion protein family named shadoo. This indicates that this region has gained an important, hitherto unknown physiological function during the evolution of the prion protein.
PrPSc
Both PrPSc and PrPC are encoded by the same host gene. Although the amino acid sequence of PrPSc and PrPC are identical the proteins differ from each other in their tertiary structure and their biochemical and physicochemical properties. Low and high resolution structural methods showed that PrPC is rich in α-helical secondary structure. To date, no structural data at the atomic level exist for PrPSc, but optical measurements and Fourier-transform infrared spectroscopy showed an increase in β-sheet structure for PrPSc when compared to PrPC.
In view of their physicochemical differences, PrPC is monomeric and soluble in nondenaturing detergents, whereas PrPSc forms oligomers and aggregates that cannot be dissolved in detergents. Another characteristic is that PrPC is sensitive to Proteinase K digestion, whilst PrPSc can only be partially degraded by Proteinase K leading to an N-terminally truncated resistant core with a size of 27–30 kDa. This fragment is termed PrP27-30. Diglycosylated, GPI-anchored PrPSc has a size of 33–35 kDa as judged by its electrophoretic mobility on a SDS-PAGE.
Nucleic acid
No prion-specific nucleic acid has been detected.
Proteins
The three-dimensional structures of recombinant PrP from a wide variety of species have been solved by nuclear magnetic resonance spectroscopy in solution. The overall architecture of the proteins is nearly identical containing an N-terminally flexible tail of residues 23–125 and a globular folded C-terminal domain of residues 126–231. The C-terminal domain is an autonomously folding unit and contains three α-helices and a short antiparallel β-sheet of residues 128–131 and 161–164 (Figure 1B). A comparison of the recombinant protein with PrPC purified from calf brain showed that the fold of the recombinant prion proteins is identical with the natural cellular protein and that the posttranslational modifications of PrPC do not affect the fold.
More recently, a special structural feature was identified in the loop β2–α2 between the second β-sheet and the second α-helix showing high conformational variability between species. Whereas the loop β2–α2 is flexible disordered in the prion proteins of most species, it is extremely well-ordered in the prion proteins from elk, bank vole and tammar wallaby.
In addition, crystal structures have been determined for the C-terminal domain in complex with monoclonal antibodies or FAB fragments, indicating a similar fold for PrPC as obtained by NMR spectroscopy. The only exception represents the crystal structure of a domain-swapped dimer in which the third α-helices are swapped and the disulfide bridge is newly arranged between the two molecules.
Morphology
High resolution structural studies using X-ray crystallography and solution NMR spectroscopy for PrPSc are more challenging due to the limited solubility of PrPSc. First morphological characterizations for PrP were obtained from rod-shaped particles that were isolated from detergent and proteinase K-treated microsomal fractions which were enriched for prion infectivity. These rod-shaped particles have a width of 10–20 nm and mean lengths of 100–200 nm. The rods are smooth, almost ribbon-like, and infrequently are twisted. The fine structure of these aggregates was originally designated by Mertz as scrapie-associated fibrils (SAF). These partially purified rods show the ultrastructural and tinctorial properties of amyloid, including Thioflavin T positivity and the green–gold birefringence obtained under cross-polarized light after Congo Red staining, indicating the presence of cross β-sheet secondary structure. The purified fractions of the rods also contained 2D crystals, which were used for crystallographic studies and structural models for PrPSc based on these data were created.
Several in vitro conversion model systems to obtain PrP fibers from prion proteins produced in Escherichia coli were also used to study their morphological and structural aspects. Amyloid fibers obtained with these assays under physiological conditions appear as long and unbranched fibers in negatively stained electron micrographs (Figure 1C). Low resolution structural data are also available for recombinant PrP and PrP fragments.
Soluble oligomers rather than insoluble fibrils have been hypothesized in other amyloid diseases such as Alzheimer`s disease to be the relevant toxic species and the formation of large aggregates might have a protective effect by sequestering the more toxic oligomers. Solubilization of PrP27-30 into liposomes indicates that large fibrillar structures are not required for infectivity and that PrPSc oligomers are the relevant molecules for infectivity. To investigate the relationship between size and infectivity in prion diseases, purified PrPres was partially disaggregated to obtain a spectrum of different-sized aggregates. It turned out that particles with a size of 17–27 nm which corresponds to a molecular weight of 300–600 kDa and an association of 14–28 PrP molecules are the most infectious particles. These particles are non-fibrillar and appear as small amorphous ellipsoidal and spherical particles with a diameter of 20–25 nm in transmission electron micrographs.
Lipids
The prion protein is attached by a glycosylphosphatidylinositol anchor (GPI) in cholesterol rich lipid rafts in the outer leaflet of the cell membrane of neurons to its carboxy-terminal serine residue. The glycan core of the GPI-anchor consists of mannose-mannose-mannose-(N-acetyl neuraminic acid-galactose-N-acetylgalactosamine)-mannose-glucosamine-myo-inositol. As commonly found for GPI anchored proteins, this core is attached to the cell membrane by a phospholipid membrane and to the C-terminus of the GPI-anchored protein through a phosphoethanolamine bridge.
PrPC can be released from the cell surface by digestion with phosphoinositide-specific phospholipase C (PI-PLC), whereas PrPSc cannot be released by the enzyme, which indicates that the cleavage site is not accessible due to the conformational rearrangement of PrPC. The importance of the GPI anchor for disease was recently demonstrated by the generation of transgenic mice that express a PrP construct lacking the GPI anchor. After infection with RML prions these heterozygous mice developed widely spread plaques in the brain, but without developing any clinical signs of disease. In a follow-up study the authors reported that infection of homozygous transgenic mice expressing anchorless PrP with RML induced a new fatal disease with unique clinical signs and altered neuropathology.
Carbohydrates
PrP also contains two highly conserved Asn-Xaa-Ser/Thr sequons for N-linked glycosylation at Asn-181 and Asn-197 (numeration according to human PrP, resulting in di-, mono- and unglycosylated states of the protein. Mass spectrometry analysis of the two N-linked oligosaccharides of PrPC and PrPSc from hamster and mouse showed that the oligosaccharides consist of different neutral and sialylated bi-, tri- and tetra-antennary oligosaccharides with more complex acidic glycan structures at Asn-196. In addition, a higher content of tri- and tetra-antennary glycans and reduced levels of glycans with bisecting GlcNAc residues were identified for PrPSc, leading to the suggestion that the activity of N-acetylglucosaminyltransferase III is reduced. No O-linked glycosylation has been identified.
Genome organization and replication
Genome organization
The PrP gene has been identified in many mammalian species, birds, reptiles and amphibians. PrP-like genes have also been identified in fish. The gene encoding the prion protein consists of three exons, from which exon 3 encodes the complete mature protein. The entire ORF of all known mammalian PrP genes is contained within a single exon. The gene encoding the human prion protein is located on chromosome 20 in a single exon, whereas the gene encoding the murine prion protein is located on the systemic chromosome 2.
In general, PrP genes are composed of three exons, as clearly demonstrated for mouse and sheep. The PrP genes of humans and Syrian hamsters (SHa) appear to have three exons but most HuPrP and SHaPrP mRNAs are spliced from only two exons that are separated by ~10 kb. Exon-1 of the SHaPrP gene encodes a portion of the 5′ untranslated leader sequence while exon-3 encodes the ORF and the 3′-UTR. The ORF of the HuPrP gene encodes a protein of 253 amino acids while the mouse and SHaPrP genes encode proteins of 254 residues. The promoters of the PrP genes of both animals contain three or two repeats, respectively, of G–C nonamers, but are devoid of TATA boxes. These nonamers represent a motif which may function as a canonical binding site for transcription factor Sp1.
The sequence similarity between the ORFs encoding different mammalian PrPs is ~90%: 30–50% identities and around 50% similarities are found between mammals, birds, reptiles and amphibians. The sequence homology between fish and tetrapod PrPs is less than 25%.
Replication
Several hypotheses have been described to explain the unusual nature of the infectious prion agent. The most widely discussed hypotheses are (i) the “protein-only” hypothesis, (ii) the virino hypothesis and (iii) the hypothesis that stoichometric transformation of PrPC to PrPScin vitro requires specific RNA molecules.
Although the “protein-only” hypothesis has long been under debate, it is to date the most widely accepted theory to explain the unusual nature of the infectious agent. The “protein-only” hypothesis postulates that a protein rather than a virus or nucleic acid is the infectious agent which transfers the disease related information from one host to another. The “protein-only” hypothesis was first formulated by Alper and Griffith in 1967, renewed by Prusiner in 1982 and later refined by Weissmann. It postulates that the infectious agent consists solely of a host-encoded protein. The propagation is assumed to be a post-translational conformational process in which the infectious agent causes the refolding of the healthy form of the prion protein into the pathogenic isoform by acting as a template. An increasing number of experiments are in line with the “protein-only” hypothesis. The first strong argument is that prions cannot be transmitted to PrP knockout mice in which the Prnp gene is removed and so these mice do not carry prion infectivity in the brain. Mice overexpressing PrPC were found to be highly susceptible to scrapie. Futhermore, familial human prion diseases are associated with mutations in the PRNP gene. This group of experiments strongly indicates that either PrPC or mutations in the gene encoding PrP are needed to develop a prion disease.
Over the past years many efforts have been undertaken to prove the “protein-only” hypothesis by the de novo generation of infectious prions from either noninfectious brain-derived PrPC or recombinant PrP produced in E. coli. The first evidence consisted of producing partially proteinase K resistant PrP (PrPres) from radioactivity labeled PrPC in cell-free systems. However, the amount of the de novo produced PrPSc was low compared to the starting material and was not infectious.
Further support for the hypothesis was obtained from several studies using the PMCA (protein misfolding cyclic amplification) technology. This assay in which PrPres is multiplied by serial amplification steps was developed by Soto and colleagues. In the PMCA, brain homogenate from healthy animals is mixed with small quantities of PrPres. After several rounds of incubation, sonicating and dilution with fresh brain homogenate from healthy animals, PrPres can be amplified in large quantities. In one approach, PMCA generated hamster PrPres could induce scrapie in wild-type hamster. Modified protocols of the PMCA in which either purified PrPC or recombinant expressed PrP have been used as substrate were also applied. However, the sensitivity was much lower than in the PMCA performed with brain homogenate, suggesting that additional auxiliary factors might be necessary for successful amplification.
A systematic investigation for the minimal compounds needed for efficient amplification was performed by Supattapone and coworkers. In this approach PrPres was produced from purified PrPC, copurified lipids and single stranded polyanions. Inoculation of these de novo prions caused TSE in wild-type hamster. The most convincing evidence for the “protein-only” hypothesis comes from a study that was recently published by Ma and coworkers. Here, infectious prions were obtained with the PMCA using PrPC purified from E. coli extracts, anionic phospholipids (POPG) and RNA obtained from mouse liver. These prions induced a neurologic disease in wild-type mice after 130 days with the classical signs of a transmissible prion disease. In other approaches, recombinant prion proteins without any cofactors were converted into cross-β-sheet aggregates that are capable of inducing a transmissible prion disease in transgenic mice or wild-type hamster.
Genetic evidence that supports the “protein-only” hypothesis was obtained from a study in which transgenic mice expressing two amino acids at positions 170 and 174 from the elk prion protein in the Prnp gene were generated. These two mutations lead to a well-structured loop that connects the second β-sheet and the second a-helix in the C-terminal domain of the protein. Mouse lines expressing these transgenes develop a spontaneous prion disease which is transmissible to transgenic mice overexpressing wild-type PrP and consecutively to wild-type mice.
Antigenic properties
The generation of antibodies against PrPC and PrPSc is difficult, because of the strong self-tolerance of PrPC. These limitations have been overcome by using Prnp0/0 mice, which are not immune tolerant to PrPC, and show a strong immune response against PrPC. These mice have been used for the production of highly efficient monoclonal antibodies, including the “POM” library, which contains antibodies directed against conformational epitopes in the folded C-terminal part of the protein as well as against linear epitopes in the unstructured N-terminal part. These monoclonal antibodies represent valuable high immunoaffinity reagents that can be applied in a variety of techniques for the detection of PrPC and PrPSc.
Several studies showed that anti-PrP antibodies have neutralizing effects by clearing PrPSc levels in scrapie-infected cell lines, in cell-free systems and by recognition of PrPC in transgenic mice on intraperitoneal prion inoculation. Furthermore, passive transfer of anti-PrP monoclonal antibodies delays the onset of scrapie in mice infected with prions by the intraperitoneal route. However, this method requires high levels of the antibodies to cause a significant effect on the survival of prion-infected mice and it has been ineffective in mice showing already clinical signs, which might be due to the blood–brain barrier (BBB) impermeability of the antibodies. Active immunization as antibody-based anti-prion prophylaxis is still difficult to achieve, although intense effort has been made to circumvent the tolerance of the humoral immune system to PrPC by using different antigens and various antibody retrieval techniques.
The production of antibodies directed against PrPSc is more challenging as this requires binding of the antibodies against specific conformational epitopes that are only exposed in PrPSc. Antibodies that specifically bind PrPSc without binding to PrPC have been reported, but their affinity seems to be poor and their diagnostic value has awaited confirmation for more than a decade.
Biological properties
Physiological function of PrPC
Despite extensive research, the physiological function of PrPC is still unclear. A clarification of the function of PrPC was expected from the generation of PrPC knock out mice. However, these transgenic mouse lines show only a subtle phenotype. Since then several biological functions, some of which are controversial, have been attributed to PrPC, including neuronal signal transduction, lymphocyte function, copper-binding, as well as pro- and anti-apoptotic functions.
In the search for intrinsic functional domains of PrPC several mouse lines were generated with specific deletions in the PrnP gene. These mice developed several prionopathies characterized by shortened life span and the development of white matter disease in the central nervous system (CNS) as well as neuronal death in the cerebellum.
More recently, PrPC was shown to play an important role in maintaining peripheral myelin in Schwann cells, indicating that PrPC acts as a signaling protein in axons necessary to keep peripheral nerves intact. The absence of PrPC causes otherwise demyelination in the Schwann cells.
Mutations in the human PrP genes
About 10–15% of the CJD cases and virtually all cases of GSS and FFI are autosomal dominantly inherited prion diseases with point mutations, amino acid insertions or deletions in the PRNP gene (Table 3). There is variability in the clinical and pathological findings, the age of onset and the duration depending on the particular PRNP mutation involved. Some PRNP mutations might cause neurodegenerative diseases that are not transmissible and therefore represent a proteinopathy rather than a prion disease.
Most of the point mutations are located in the well-structured C-terminal domain in PrPC (Table 3; Figure 2A–C) and in the immediate preceding fragment of residues 102–125. Amino acid insertions and deletions are found in the N-terminal part of the protein affecting the length of the octapeptide repeat region.
Table 3 Human PrP gene mutations found in the inherited prion diseases
| Inherited prion disease | PrP gene mutation |
| Familial Creutzfeldt–Jakob disease | D178N–129V, V180I, V180I–M232R, T183A, T188A, E196K, E200K, V203I, E208H, V210I, E211Q, M232R |
| Gerstmann–Sträussler–Scheinker syndrome | P102L-129M, P105L–129V, A117V–129V, G131V–129M, F198S–129V, D202N–129V, Q212P, Q217R–129M, M232T |
| Fatal familial insomnia | D178N-129M |
| Mutations in PRNP associated with familial dementia and/or neuropsychiatric symptoms PrP (Not further classified) | I138M, G142S, Q160Stop–129M, T188K, T188R, M232R, P238S |
| Vascular amyloid depositions | Y145STOP-129M |
| Base pair insertion/deletion | Insertions: 24 bp, 48 bp, 96 bp, 120 bp, 144 bp, 168 bp, 192 bp, 216 bp; Deletion: 24 bp |
|
|
|
| Proven, but unclassified prion disease | H187R |
| Polymorphisms | N171S, E219K |
| Silent polymorphisms | P68P, A117A, G124G, V161V, N173N, H177H, T188T, D202D, Q212Q, R228R, S230S |
Point mutations, insertions and deletions identified in the human PRND gene in patients. The polymorphism at amino acid position 129 that is associated with disease is also indicated. Octarepeat inserts are designated as bpi (base pair insertion); Octarepeat deletions as bpd (base pair deletion).
Table based on Aguzzi and Calella (2009). Physiol. Rev., 89, 1105-1152; Aguzzi, Baumann and Bremer (2008). Annu. Rev. Neurosci., 31, 439–477.
Polymorphisms in PrP genes
The M/V polymorphism at codon 129 in HuPrP influences the incidence of sporadic and infectious CJD as well as the age of onset of inherited prion diseases. Homozygosity at codon 129 is more frequent in both sporadic and infectious CJD; it also heralds an earlier age of clinical disease in people carrying pathologic mutations. Those individuals who carry the DI78N mutation develop FFI manifested by insomnia and dysautonomia if they encode M129 on the mutant allele, while those that encode V129 present with fCJD are characterized by dementia (Table 3).
The E/K polymorphism at codon 219 in HuPrP seems to influence resistance to CJD. Individuals with wild-type PrP sequences who are heterozygous for K219 appear to be resistant to CJD. This dominant negative effect is likely to be the results of increased affinity of PrPC (K219) for protein X which is thought to function like a molecular chaperone in facilitating the conversion of PrPC into PrPSc.
The Q/R polymorphism at codon 171 in OvPrP seems to govern the resistance of sheep to natural scrapie. Sheep with wild-type PrP sequences that are heterozygous for R171 appear to be resistant to natural scrapie, but scrapie can be transmitted to some of these animals by inoculation of prions. This dominant negative effect is also likely to be the result of increased affinity of PrPC (R171) for protein X. A polymorphism at codon 136 is also thought to influence the susceptibility of some breeds of sheep to scrapie.
The S/F polymorphism at codon 225 in mule deer influences susceptibility to CWD. In addition, elk have a polymorphism at codon 132 (M/L) of PrnP corresponding to the polymorphic codon 129 (M/V) in humans. Elk with codon 132LL experimentally infected with CWD were resistant to infection for at least 4 years, whereas 132MM or 132ML elk developed terminally clinical disease after 23 or 40 months. A reduced susceptibility to CWD was reported in white-tailed deer with G/S and Q/H polymorphisms at codons 96 and 95, respectively.
Prion strains
One of the most intriguing phenomena in prion diseases is the appearance of different strains. Prion strains are defined as infectious isolates that, when transmitted to identical hosts, exhibit distinct disease phenotypes. Prion strains can be discriminated by specific incubation times, histopathological lesion profiling and clinical signs. Most phenotypes of strains are relatively stable during serial transmission. Often distinct new strains are observed upon transmission of prions across a transmission barrier or into animals that express specific polymorphisms of the prion gene, a phenomenon sometimes referred to as a “strain mutation”. The transmission barrier is explained by differences in the amino acid sequence between two species whereby single amino acid changes can lead to significant changes in the incubation times.
To explain the appearance of different prion strains within the framework of the “protein-only” hypothesis it was suggested that the information for the strain-specific characteristics and phenotypes must be enciphered in the PrPSc conformation itself or in other words PrPSc of each strain exhibits a different conformation. A possible molecular explanation for the appearance of different conformations was recently provided from a systematic structural study on fibril-forming peptides derived from prion and other amyloid proteins. These peptides form small β-sheets and adopt distinct crystalline polymorphisms according to their relative orientation and packing arrangements. Each different crystalline polymorph could thereby retain and transfer the strain-specific information from one species to another. Experimental evidence for strain-specific PrPSc conformations include the different electrophoretic mobilities of different strains, the immunological reactivity to amino-proximal antibodies upon proteinase K digestion and the different susceptibilities of PrPSc derived from different strains to chaotropic salts. In addition, strains can be discriminated by their accessibility of specific epitopes to monoclonal antibodies. These epitopes are fully accessible in PrPC, but buried in PrPSc and only become exposed after treatment with defined concentrations of chaotropic salts.
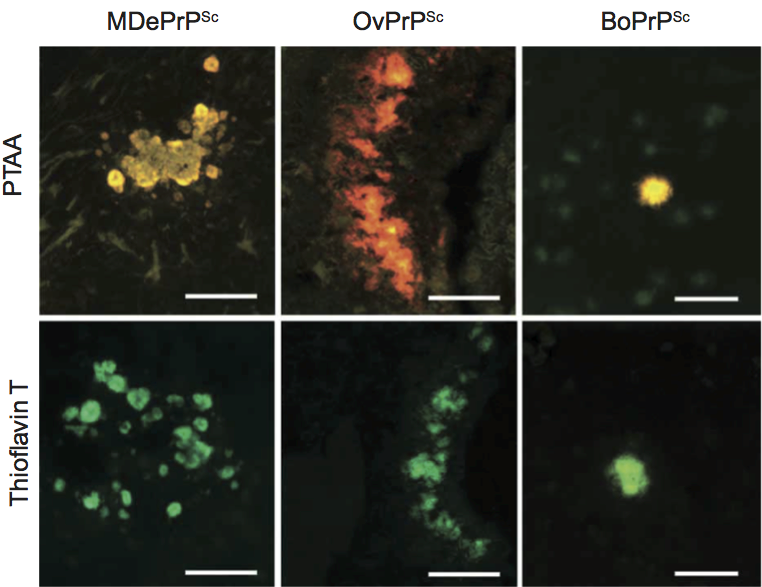
Optical tools to classify cross-β-sheet amyloid deposits are historically small amyloidotropic dyes such as Congo red and Thioflavin T. However, the use of these dyes is limited to the detection of total amyloid per se and different amyloid morphologies as present in prion strains cannot be separated with these dyes. More sophisticated tools would therefore be of high interest for the diagnosis of amyloid deposits with distinct morphologies. Luminescent conjugated polymers (LCPs) are a class of novel amyloidotropic dyes that have recently been proven to overcome these limitations. LCPs are small fluorescent probes that were originally synthesized for use in electrical devices. These dyes exhibit a highly flexible backbone that adopts a distinct conformation upon binding to the ordered structure of amyloid fibrils and generates a fluorescent signature that is specific for the structural symmetry of these fibrils. The spectral properties of the LCPs were used for the analysis of brain sections from mice that were infected with distinct prion strains (Figure 3). The LCPs could discriminate different prion strains due to individual staining patterns and could even detect prion deposits that were negative for Congo red and Thioflavin T. These findings provide additional evidence that PrPSc strains might be enciphered in the conformation of the prion aggregate.
Taxonomic considerations of mammalian prions
A listing of the different mammalian prions is given in Table 1. Although the prions that cause TME and BSE are referred to as TME prions and BSE prions; this may be unjustified, because both are thought to originate from the oral consumption of scrapie prions in sheep-derived foodstuffs and because many lines of evidence argue that the only difference among the various prions is the sequence of PrP which is dictated by the host and not the prion itself. The human prions present a similar semantic conundrum. Transmission of human prions to laboratory animals produces prions carrying PrP molecules with sequences dictated by the PrP gene of the host, not that of the inoculum.
To simplify the terminology, the generic term PrPSc is suggested in place of such terms as PrPCJD, PrPBSE and PrPres. To distinguish PrPSc found in humans or cattle from that found in other animals, we suggest HuPrPSc or BoPrPSc instead of PrPCJD or PrPBSE, respectively (Table 1). Once human prions and thus HuPrPSc molecules have been passaged into animals, then the prions and PrPSc are no longer of the human species unless they were formed in an animal expressing a HuPrP transgene. In the case of mutant PrPs, the mutation and any important polymorphism can be denoted in parentheses following the particular PrP isoform. For example, in FFI, the pathogenic PrP isoform would be referred to as PrPSc or HuPrPSc; alternatively, if it were important to identify the mutation, then it would be written as HuPrPSc (D178N, M129) (Table 3). The term PrPres or PrP-res is derived from the protease-resistance of PrPSc but protease-resistance, insolubility, and high β-sheet content should be only considered as surrogate markers of PrPSc since one or more of these may not always be present. Whether PrPres is useful in denoting PrP molecules that have been subjected to procedures that modify their resistance to proteolysis, but have not been demonstrated to convey infectivity or to cause disease remains debatable.
The term PrP* has been used in two different ways. First, it has been used to identify a fraction of PrPSc molecules that are infectious. Such a designation is thought to be useful since there are ~105 PrPSc molecules per infectious unit. Second, PrP* has been used to designate a metastable intermediate of PrPC that is bound to protein X. It is noteworthy that neither a subset of biologically active PrPSc molecules nor a metastable intermediate of PrPC has been identified, to date.
In mice, the PrP gene denoted Prnp is now known to be identical with two genes denoted Sinc and Prn-i that control the length of the incubation time in mice inoculated with prions. These findings permit a welcome simplification. A gene designated Pid-1 on mouse chromosome 17 also appears to influence experimental CJD and scrapie incubation times but information on this locus is limited.
Distinguishing among CJD, GSS and FFI has grown increasingly difficult with the recognition that CJD, GSS and FFI are autosomal dominant diseases caused by mutations in the PRNP gene. Initially, it was thought that a specific PrP mutation was associated with a particular clinico-neuropathologic phenotype but an increasing number of exceptions are being recognized. Multiple examples of variations in the clinico-neuropathologic phenotype within a single family where all affected members carry the same PrP mutation have been recorded. Most patients with a PrP mutation at codon 102 present with ataxia and have PrP amyloid plaques; such patients are generally given the diagnosis of GSS, but some individuals within these families present with dementia, a clinical characteristic that is usually associated with CJD. One suggestion is to label these inherited disorders as “prion disease” followed by the mutation in parenthesis while another is the use the terms fCJD and GSS followed by the mutation. In the case of FFI, describing the Dl78N mutation and M129 polymorphism seems unnecessary since this is the only known mutation-polymorphism combination that gives the FFI phenotype.
Derivation of name
Prion: sigla from proteinaceous and infectious particle.
PrP orthologs: Doppel and Shadoo
Two other proteins, the Doppel and Shadoo, have been identified that belong to the PrP family. In contrast to PrP, these two proteins are not infectious. The Doppel protein (a German synonym of double; Dpl) was originally identified by the observation of phenotypic discrepancies in PrP0/0 mice. Two mouse lines, ZHI-Prnp0/0 mice and Edbg Prnp−/− mice, showed no obvious phenotype, whereas ZHII-Prnp0/0, Ngsk-Prnp0/0 and Rcm0-Prnp0/0 developed late onset ataxia caused by cerebellar Purkinje cell degeneration. The identification of a novel gene locus (Prnd) 16 kb downstream of the Prnp gene and its product, Dpl, provided an explanation for the different phenotypes.
The amino acid identity between PrP and Dpl is about 20%. The human Dpl consists of 179 amino acids and contains two glycosylation sites. Compared to PrP, Dpl lacks the octarepeats, the charged clusters and the hydrophobic stretch and contains an additional disulfide bridge. The N-terminal tail, residues 24–52, of Dpl is significantly shorter due to the lack of the octarepeats, but the C-terminal domain of residues 52–149 has a similar structural organization as seen for PrP. The well-structured C-terminal domain of the human Dpl contains four α-helices comprising residues 72–80 (α1), 101–115 (α2a), 117–121 (α2b) and 127–141 (α3) and a short antiparallel β-sheet of residues 58–60 and 88–90. The two disulfide bridges in the protein are formed between Cy109–Cys143, connecting helices α2 and α3, and between Cys95–Cys148, connecting the loop β2–α2 with the C-terminal end of Dpl.
Dpl is mainly expressed in testis and heart, to some smaller extent in other peripheral organs and at a very low expression rate in the brains of adult wild-type mice. Due to the high biochemical and structurally similarities between PrP and Dpl it was assumed that the proteins also exhibit similar physiological functions.
To elucidate the biological function of Dpl, Prnd knockout mice were generated by homozygous targeted disruption of the Prnd gene. The Prnd0/0 mice survive until adulthood like the Prnp knockout mice. In contrast to the PrP0/0, the Prnd0/0mice show a specific phenotype. Whereas females lacking Dpl were viable and fertile, males without Dpl suffer from male sterility. In addition, the Prnd gene was disrupted in the ataxic Zurich II mice by using transallelic targeted meiotic recombination to generate Prnp/Prnd double deletion mice. These mice did not show any ataxia, proving the causal role for Dpl overexpression in the Zurich II phenotype.
The PrP-like protein Shadoo (Sho) (for shadow of the prion protein) was identified in 2003 by intense database search for nucleotide sequences that are similar to PrnP. Shadoo has been shown to present in many mammals and fish. The related gene Sprn is located on chromosomes 7 and 10 in mice and humans, respectively, which are distinct from Prnp and Prnd, indicating that Sprn has evolved separately. The entire open reading frame of Sprn is contained in a single exon. Sho seems to be expressed in the central nervous system, and to be neuroprotective by preventing cerebellar degeneration mediated by CNS-expressed Dpl and N-terminal deletion mutants of PrP.
The mature protein consists of 98 amino acids and is presumably gycosylated at one site. Similarly to PrP, Sho contains the highly conserved valine- and alanine-rich hydrophobic stretch, the N-terminal signal sequence for targeting the protein into the endoplasmatic reticulum, and the GPI anchor. On the other hand, it lacks the octarepeats and has no cysteine residues. In addition, Shadoo does not contain a C-terminal domain and far-UV CD spectra indicate that the protein is unfolded.
Further reading
Note: A version of this text with full cited references is available online on ScienceDirect®, www.sciencedirect.com
Aguzzi and Calella, 2009 A. Aguzzi, A.M. Calella, Prions: protein aggregation and infectious diseases. Physiol. Rev. 89 (2009) 1105–1152.
Aguzzi and Polymenidou, 2004 A. Aguzzi, M. Polymenidou, Mammalian prion biology: one century of evolving concepts. Cell. 116 (2004) 313–327.
Aguzzi et al., 2008 A. Aguzzi, F. Baumann, J. Bremer, The prion's elusive reason for being. Annu. Rev. Neurosci. 31 (2008) 439–477.
Aguzzi et al., 2007 A. Aguzzi, M. Heikenwalder, M. Polymenidou, Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 8 (2007) 552–561.
Aguzzi et al., 2008 A. Aguzzi, C. Sigurdson, M. Heikenwalder, Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol. 3 (2008) 11–40.
Collinge, 1997 J. Collinge, Human prion diseases and bovine spongiform encephalopathy (BSE). Hum. Mol. Genet. 6 (1997) 1699–1705.
Falsig et al., 2008 J. Falsig, K.P. Nilsson, T.P. Knowles, A. Aguzzi, Chemical and biophysical insights into the propagation of prion strains. HFSP J. 2 (2008) 332–341.
Heppner and Aguzzi, 2004 F.L. Heppner, A. Aguzzi, Recent developments in prion immunotherapy. Curr. Opin. Immunol. 16 (2004) 594–598.
Prusiner, 1982 S.B. Prusiner, Novel proteinaceous infectious particles cause scrapie. Science. 216 (1982) 136–144.
Watts and Westaway, 2007 J.C. Watts, D. Westaway, The prion protein family: diversity, rivalry, and dysfunction. Biochim. Biophys. Acta. 1772 (2007) 654–672.
Weissmann, 1991 C. Weissmann, A “unified theory” of prion propagation. Nature. 352 (1991) 679–683.
Weissmann, 1996 C. Weissmann, The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett. 389 (1996) 3–11.
Weissmann and Aguzzi, 2005 C. Weissmann, A. Aguzzi, Approaches to therapy of prion diseases. Annu. Rev. Med. 56 (2005) 321–344.
Weissmann and Flechsig, 2003 C. Weissmann, E. Flechsig, PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 6 (2003) 43–60.
References
1. Gordon, W. S. (1946) Vet Rec 58, 516-525
2. Heikenwalder, M., Zeller, N., Seeger, H., Prinz, M., Klohn, P. C., Schwarz, P., Ruddle, N. H., Weissmann, C., and Aguzzi, A. (2005) Science 307, 1107-1110
3. Ligios, C., Sigurdson, C. J., Santucciu, C., Carcassola, G., Manco, G., Basagni, M., Maestrale, C., Cancedda, M. G., Madau, L., and Aguzzi, A. (2005) Nat Med 11, 1137-1138
4. Wilesmith, J. W., Ryan, J. B., Hueston, W. D., and Hoinville, L. J. (1992) Vet Rec 130, 90-94
5. Williams, E. S., and Miller, M. W. (2002) Rev Sci Tech 21, 305-316
6. Williams, E. S. (2005) Vet Pathol 42, 530-549
7. Sigurdson, C. J., and Aguzzi, A. (2007) Biochim Biophys Acta 1772, 610-618
8. Miller, M. W., Williams, E. S., McCarty, C. W., Spraker, T. R., Kreeger, T. J., Larsen, C. T., and Thorne, E. T. (2000) J Wildl Dis 36, 676-690
9. Natl. Wildl. Health Cent. Map of chronic wating disease in North America. http://www.nwhc.usgs.gov/disease_information/chronic_wasting_disease/no…
10. Williams, E. S., and Young, S. (1982) J Wildl Dis 18, 465-471
11. Baeten, L. A., Powers, B. E., Jewell, J. E., Spraker, T. R., and Miller, M. W. (2007) J Wildl Dis 43, 309-314
12. Williams, E. S., and Young, S. (1980) J Wildl Dis 16, 89-98
13. Miller, M. W., and Williams, E. S. (2003) Nature 425, 35-36
14. Weissmann, C., and Aguzzi, A. (2005) Annu Rev Med 56, 321-344
15. Harries-Jones, R., Knight, R., Will, R. G., Cousens, S., Smith, P. G., and Matthews, W. B. (1988) J Neurol Neurosurg Psychiatry 51, 1113-1119
16. Weissmann, C., and Aguzzi, A. (1997) Curr Opin Neurobiol 7, 695-700
17. Collinge, J. (1997) Hum Mol Genet 6, 1699-1705
18. Aguzzi, A., and Polymenidou, M. (2004) Cell 116, 313-327
19. Will, R. G., Ironside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K., Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., and Smith, P. G. (1996) Lancet 347, 921-925
20. Hill, A. F., Desbruslais, M., Joiner, S., Sidle, K. C., Gowland, I., Collinge, J., Doey, L. J., and Lantos, P. (1997) Nature 389, 448-450, 526
21. Bruce, M. E., Will, R. G., Ironside, J. W., McConnell, I., Drummond, D., Suttie, A., McCardle, L., Chree, A., Hope, J., Birkett, C., Cousens, S., Fraser, H., and Bostock, C. J. (1997) Nature 389, 498-501
22. Gajdusek, D. C. (1977) Science 197, 943-960
23. Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987) Cell 51, 229-240
24. Meyer, R. K., McKinley, M. P., Bowman, K. A., Braunfeld, M. B., Barry, R. A., and Prusiner, S. B. (1986) Proc Natl Acad Sci U S A 83, 2310-2314
25. Stahl, N., and Prusiner, S. B. (1991) FASEB J 5, 2799-2807
26. Bolton, D. C., Meyer, R. K., and Prusiner, S. B. (1985) J Virol 53, 596-606
27. Haraguchi, T., Fisher, S., Olofsson, S., Endo, T., Groth, D., Tarentino, A., Borchelt, D. R., Teplow, D., Hood, L., Burlingame, A., and et al. (1989) Arch Biochem Biophys 274, 1-13
28. Basler, K., Oesch, B., Scott, M., Westaway, D., Walchli, M., Groth, D. F., McKinley, M. P., Prusiner, S. B., and Weissmann, C. (1986) Cell 46, 417-428
29. Pan, K. M., Stahl, N., and Prusiner, S. B. (1992) Protein Sci 1, 1343-1352
30. Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wüthrich, K. (1996) Nature 382, 180-182
31. Safar, J., Roller, P. P., Gajdusek, D. C., and Gibbs, C. J., Jr. (1993) J Biol Chem 268, 20276-20284
32. James, T. L., Liu, H., Ulyanov, N. B., Farr-Jones, S., Zhang, H., Donne, D. G., Kaneko, K., Groth, D., Mehlhorn, I., Prusiner, S. B., and Cohen, F. E. (1997) Proc Natl Acad Sci U S A 94, 10086-10091
33. Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J., Cohen, F. E., and et al. (1993) Proc Natl Acad Sci U S A 90, 10962-10966
34. Caughey, B. W., Dong, A., Bhat, K. S., Ernst, D., Hayes, S. F., and Caughey, W. S. (1991) Biochemistry 30, 7672-7680
35. Oesch, B., Westaway, D., Walchli, M., McKinley, M. P., Kent, S. B., Aebersold, R., Barry, R. A., Tempst, P., Teplow, D. B., Hood, L. E., and et al. (1985) Cell 40, 735-746.
36. Liu, H., Farr-Jones, S., Ulyanov, N. B., Llinas, M., Marqusee, S., Groth, D., Cohen, F. E., Prusiner, S. B., and James, T. L. (1999) Biochemistry 38, 5362-5377
37. López García, F., Zahn, R., Riek, R., and Wüthrich, K. (2000) Proc Natl Acad Sci U S A 97, 8334-8339
38. Riek, R., Hornemann, S., Wider, G., Glockshuber, R., and Wüthrich, K. (1997) FEBS Lett 413, 282-288
39. Lysek, D. A., Schorn, C., Nivon, L. G., Esteve-Moya, V., Christen, B., Calzolai, L., von Schroetter, C., Fiorito, F., Herrmann, T., Güntert, P., and Wüthrich, K. (2005) Proc Natl Acad Sci U S A 102, 640-645
40. Christen, B., Pérez, D. R., Hornemann, S., and Wüthrich, K. (2008) J Mol Biol 383, 306-312
41. Christen, B., Hornemann, S., Damberger, F. F., and Wüthrich, K. (2009) J Mol Biol 389, 833-845
42. Zahn, R., Liu, A., Lührs, T., Riek, R., von Schroetter, C., López García, F., Billeter, M., Calzolai, L., Wider, G., and Wüthrich, K. (2000) Proc Natl Acad Sci U S A 97, 145-150
43. Gossert, A. D., Bonjour, S., Lysek, D. A., Fiorito, F., and Wüthrich, K. (2005) Proc Natl Acad Sci U S A 102, 646-650
44. Hornemann, S., and Glockshuber, R. (1996) J Mol Biol 261, 614-619
45. Hornemann, S., Schorn, C., and Wüthrich, K. (2004) EMBO Rep 5, 1159-1164
46. Antonyuk, S. V., Trevitt, C. R., Strange, R. W., Jackson, G. S., Sangar, D., Batchelor, M., Cooper, S., Fraser, C., Jones, S., Georgiou, T., Khalili-Shirazi, A., Clarke, A. R., Hasnain, S. S., and Collinge, J. (2009) Proc Natl Acad Sci U S A 106, 2554-2558
47. Haire, L. F., Whyte, S. M., Vasisht, N., Gill, A. C., Verma, C., Dodson, E. J., Dodson, G. G., and Bayley, P. M. (2004) J Mol Biol 336, 1175-1183
48. Eghiaian, F., Grosclaude, J., Lesceu, S., Debey, P., Doublet, B., Treguer, E., Rezaei, H., and Knossow, M. (2004) Proc Natl Acad Sci U S A 101, 10254-10259
49. Silveira, J. R., Raymond, G. J., Hughson, A. G., Race, R. E., Sim, V. L., Hayes, S. F., and Caughey, B. (2005) Nature 437, 257-261
50. Knaus, K. J., Morillas, M., Swietnicki, W., Malone, M., Surewicz, W. K., and Yee, V. C. (2001) Nat Struct Biol 8, 770-774
51. McKinley, M. P., Meyer, R. K., Kenaga, L., Rahbar, F., Cotter, R., Serban, A., and Prusiner, S. B. (1991) J Virol 65, 1340-1351
52. Prusiner, S. B., McKinley, M. P., Bowman, K. A., Bolton, D. C., Bendheim, P. E., Groth, D. F., and Glenner, G. G. (1983) Cell 35, 349-358
53. Prusiner, S. B., Bolton, D. C., Groth, D. F., Bowman, K. A., Cochran, S. P., and McKinley, M. P. (1982) Biochemistry 21, 6942-6950
54. McKinley, M. P., Braunfeld, M. B., Bellinger, C. G., and Prusiner, S. B. (1986) J Infect Dis 154, 110-120
55. Merz, P. A., Somerville, R. A., Wisniewski, H. M., Manuelidis, L., and Manuelidis, E. E. (1983) Nature 306, 474-476
56. Wille, H., Michelitsch, M. D., Guenebaut, V., Supattapone, S., Serban, A., Cohen, F. E., Agard, D. A., and Prusiner, S. B. (2002) Proc Natl Acad Sci U S A 99, 3563-3568
57. Govaerts, C., Wille, H., Prusiner, S. B., and Cohen, F. E. (2004) Proc Natl Acad Sci U S A 101, 8342-8347
58. Wille, H., Govaerts, C., Borovinskiy, A., Latawiec, D., Downing, K. H., Cohen, F. E., and Prusiner, S. B. (2007) Arch Biochem Biophys 467, 239-248
59. Swietnicki, W., Morillas, M., Chen, S. G., Gambetti, P., and Surewicz, W. K. (2000) Biochemistry 39, 424-431.
60. Baskakov, I. V. (2004) J Biol Chem 279, 7671-7677
61. Lührs, T., Zahn, R., and Wüthrich, K. (2006) J Mol Biol 357, 833-841
62. Wille, H., Bian, W., McDonald, M., Kendall, A., Colby, D. W., Bloch, L., Ollesch, J., Borovinskiy, A. L., Cohen, F. E., Prusiner, S. B., and Stubbs, G. (2009) Proc Natl Acad Sci U S A 106, 16990-16995
63. Tattum, M. H., Cohen-Krausz, S., Thumanu, K., Wharton, C. W., Khalili-Shirazi, A., Jackson, G. S., Orlova, E. V., Collinge, J., Clarke, A. R., and Saibil, H. R. (2006) J Mol Biol 357, 975-985
64. Nguyen, J. T., Inouye, H., Baldwin, M. A., Fletterick, R. J., Cohen, F. E., Prusiner, S. B., and Kirschner, D. A. (1995) J Mol Biol 252, 412-422
65. Inouye, H., Bond, J., Baldwin, M. A., Ball, H. L., Prusiner, S. B., and Kirschner, D. A. (2000) J Mol Biol 300, 1283-1296
66. Salmona, M., Morbin, M., Massignan, T., Colombo, L., Mazzoleni, G., Capobianco, R., Diomede, L., Thaler, F., Mollica, L., Musco, G., Kourie, J. J., Bugiani, O., Sharma, D., Inouye, H., Kirschner, D. A., Forloni, G., and Tagliavini, F. (2003) J Biol Chem 278, 48146-48153
67. Aguzzi, A., and Calella, A. M. (2009) Physiol Rev 89, 1105-1152
68. Gabizon, R., McKinley, M. P., and Prusiner, S. B. (1987) Proc Natl Acad Sci U S A 84, 4017-4021
69. Vey, M., Pilkuhn, S., Wille, H., Nixon, R., DeArmond, S. J., Smart, E. J., Anderson, R. G., Taraboulos, A., and Prusiner, S. B. (1996) Proc Natl Acad Sci U S A 93, 14945-14949
70. Stahl, N., Baldwin, M. A., Hecker, R., Pan, K. M., Burlingame, A. L., and Prusiner, S. B. (1992) Biochemistry 31, 5043-5053
71. Paulick, M. G., and Bertozzi, C. R. (2008) Biochemistry 47, 6991-7000
72. Stahl, N., Borchelt, D. R., and Prusiner, S. B. (1990) Biochemistry 29, 5405-5412
73. Chesebro, B., Trifilo, M., Race, R., Meade-White, K., Teng, C., LaCasse, R., Raymond, L., Favara, C., Baron, G., Priola, S., Caughey, B., Masliah, E., and Oldstone, M. (2005) Science 308, 1435-1439
74. Chesebro, B., Race, B., Meade-White, K., Lacasse, R., Race, R., Klingeborn, M., Striebel, J., Dorward, D., McGovern, G., and Jeffrey, M. (2010) PLoS Pathog 6, e1000800
75. Schätzl, H. M., Da Costa, M., Taylor, L., Cohen, F. E., and Prusiner, S. B. (1995) J Mol Biol 245, 362-374
76. Endo, T., Groth, D., Prusiner, S. B., and Kobata, A. (1989) Biochemistry 28, 8380-8388
77. Stahl, N., Baldwin, M. A., Teplow, D. B., Hood, L., Gibson, B. W., Burlingame, A. L., and Prusiner, S. B. (1993) Biochemistry 32, 1991-2002
78. Stimson, E., Hope, J., Chong, A., and Burlingame, A. L. (1999) Biochemistry 38, 4885-4895
79. Rudd, P. M., Endo, T., Colominas, C., Groth, D., Wheeler, S. F., Harvey, D. J., Wormald, M. R., Serban, H., Prusiner, S. B., Kobata, A., and Dwek, R. A. (1999) Proc Natl Acad Sci U S A 96, 13044-13049
80. Rudd, P. M., Merry, A. H., Wormald, M. R., and Dwek, R. A. (2002) Curr Opin Struct Biol 12, 578-586
81. Premzl, M., Delbridge, M., Gready, J. E., Wilson, P., Johnson, M., Davis, J., Kuczek, E., and Marshall Graves, J. A. (2005) Gene 349, 121-134
82. Wopfner, F., Weidenhofer, G., Schneider, R., von Brunn, A., Gilch, S., Schwarz, T. F., Werner, T., and Schätzl, H. M. (1999) J Mol Biol 289, 1163-1178
83. Van Rheede, T., Smolenaars, M. M., Madsen, O., and De Jong, W. W. (2003) Mol Biol Evol 20, 111-121
84. Simonic, T., Duga, S., Strumbo, B., Asselta, R., Ceciliani, F., and Ronchi, S. (2000) FEBS Lett 469, 33-38
85. Strumbo, B., Ronchi, S., Bolis, L. C., and Simonic, T. (2001) FEBS Lett 508, 170-174
86. Rivera-Milla, E., Stuermer, C. A., and Malaga-Trillo, E. (2003) Trends Genet 19, 72-75
87. Oidtmann, B., Simon, D., Holtkamp, N., Hoffmann, R., and Baier, M. (2003) FEBS Lett 538, 96-100
88. Gabriel, J. M., Oesch, B., Kretzschmar, H., Scott, M., and Prusiner, S. B. (1992) Proc Natl Acad Sci U S A 89, 9097-9101
89. Hsiao, K., Baker, H. F., Crow, T. J., Poulter, M., Owen, F., Terwilliger, J. D., Westaway, D., Ott, J., and Prusiner, S. B. (1989) Nature 338, 342-345.
90. Westaway, D., Goodman, P. A., Mirenda, C. A., McKinley, M. P., Carlson, G. A., and Prusiner, S. B. (1987) Cell 51, 651-662
91. Liao, Y. C., Lebo, R. V., Clawson, G. A., and Smuckler, E. A. (1986) Science 233, 364-367
92. Watts, J. C., and Westaway, D. (2007) Biochim Biophys Acta 1772, 654-672
93. Lee, I. Y., Westaway, D., Smit, A. F., Wang, K., Seto, J., Chen, L., Acharya, C., Ankener, M., Baskin, D., Cooper, C., Yao, H., Prusiner, S. B., and Hood, L. E. (1998) Genome Res 8, 1022-1037
94. McKnight, S., and Tjian, R. (1986) Cell 46, 795-805
95. Harris, D. A., Falls, D. L., Johnson, F. A., and Fischbach, G. D. (1991) Proc Natl Acad Sci U S A 88, 7664-7668
96. Simonic, T., Duga, S., Strumbo, B., Asselta, R., Ceciliani, F., and Ronchi, S. (2000) FEBS Lett 469, 33-38.
97. Strumbo, B., Ronchi, S., Bolis, L. C., and Simonic, T. (2001) FEBS Lett 508, 170-174.
98. Rivera-Milla, E., Oidtmann, B., Panagiotidis, C. H., Baier, M., Sklaviadis, T., Hoffmann, R., Zhou, Y., Solis, G. P., Stuermer, C. A., and Malaga-Trillo, E. (2006) FASEB J 20, 317-319
99. Aguzzi, A., Sigurdson, C., and Heikenwalder, M. (2008) Annu Rev Pathol 3, 11-40
100. Soto, C., and Castilla, J. (2004) Nat Med 10 Suppl, S63-67
101. Alper, T., Cramp, W. A., Haig, D. A., and Clarke, M. C. (1967) Nature 214, 764-766
102. Griffith, J. S. (1967) Nature 215, 1043-1044
103. Prusiner, S. B. (1982) Science 216, 136-144
104. Weissmann, C. (1991) Nature 352, 679-683
105. Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M., and Weissmann, C. (1993) Cell 73, 1339-1347
106. Sailer, A., Bueler, H., Fischer, M., Aguzzi, A., and Weissmann, C. (1994) Cell 77, 967-968
107. Fischer, M., Rulicke, T., Raeber, A., Sailer, A., Moser, M., Oesch, B., Brandner, S., Aguzzi, A., and Weissmann, C. (1996) EMBO J 15, 1255-1264
108. Hsiao, K., Dlouhy, S. R., Farlow, M. R., Cass, C., Da Costa, M., Conneally, P. M., Hodes, M. E., Ghetti, B., and Prusiner, S. B. (1992) Nat Genet 1, 68-71
109. Aguzzi, A., Sigurdson, C., and Heikenwaelder, M. (2008) Annu Rev Pathol 3, 11-40
110. Kocisko, D. A., Come, J. H., Priola, S. A., Chesebro, B., Raymond, G. J., Lansbury, P. T., and Caughey, B. (1994) Nature 370, 471-474
111. Bessen, R. A., Raymond, G. J., and Caughey, B. (1997) J Biol Chem 272, 15227-15231
112. Weissmann, C. (1996) FEBS Lett 389, 3-11
113. Saborio, G. P., Permanne, B., and Soto, C. (2001) Nature 411, 810-813
114. Saa, P., Castilla, J., and Soto, C. (2006) J Biol Chem 281, 35245-35252
115. Castilla, J., Saa, P., Hetz, C., and Soto, C. (2005) Cell 121, 195-206
116. Deleault, N. R., Geoghegan, J. C., Nishina, K., Kascsak, R., Williamson, R. A., and Supattapone, S. (2005) J Biol Chem 280, 26873-26879
117. Atarashi, R., Moore, R. A., Sim, V. L., Hughson, A. G., Dorward, D. W., Onwubiko, H. A., Priola, S. A., and Caughey, B. (2007) Nat Methods 4, 645-650
118. Aguzzi, A. (2007) Nat Methods 4, 614-616
119. Deleault, N. R., Harris, B. T., Rees, J. R., and Supattapone, S. (2007) Proc Natl Acad Sci U S A 104, 9741-9746
120. Wang, F., Wang, X., Yuan, C. G., and Ma, J. (2010) Science 327, 1132-1135
121. Legname, G., Baskakov, I. V., Nguyen, H. O., Riesner, D., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2004) Science 305, 673-676
122. Legname, G., Nguyen, H. O., Baskakov, I. V., Cohen, F. E., Dearmond, S. J., and Prusiner, S. B. (2005) Proc Natl Acad Sci U S A 102, 2168-2173
123. Colby, D. W., Wain, R., Baskakov, I. V., Legname, G., Palmer, C. G., Nguyen, H. O., Lemus, A., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2010) PLoS Pathog 6, e1000736
124. Makarava, N., Kovacs, G. G., Bocharova, O., Savtchenko, R., Alexeeva, I., Budka, H., Rohwer, R. G., and Baskakov, I. V. (2010) Acta Neuropathol 119, 177-187
125. Sigurdson, C. J., Nilsson, K. P., Hornemann, S., Heikenwalder, M., Manco, G., Schwarz, P., Ott, D., Rulicke, T., Liberski, P. P., Julius, C., Falsig, J., Stitz, L., Wüthrich, K., and Aguzzi, A. (2009) Proc Natl Acad Sci U S A 106, 304-309
126. Büeler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., Prusiner, S. B., Aguet, M., and Weissmann, C. (1992) Nature 356, 577-582.
127. Brandner, S., Raeber, A., Sailer, A., Blattler, T., Fischer, M., Weissmann, C., and Aguzzi, A. (1996) Proc Natl Acad Sci U S A 93, 13148-13151
128. Polymenidou, M., Moos, R., Scott, M., Sigurdson, C., Shi, Y. Z., Yajima, B., Hafner-Bratkovic, I., Jerala, R., Hornemann, S., Wüthrich, K., Bellon, A., Vey, M., Garen, G., James, M. N., Kav, N., and Aguzzi, A. (2008) PLoS One 3, e3872
129. Horiuchi, M., and Caughey, B. (1999) EMBO J 18, 3193-3203
130. Enari, M., Flechsig, E., and Weissmann, C. (2001) Proc Natl Acad Sci U S A 98, 9295-9299
131. Peretz, D., Williamson, R. A., Kaneko, K., Vergara, J., Leclerc, E., Schmitt-Ulms, G., Mehlhorn, I. R., Legname, G., Wormald, M. R., Rudd, P. M., Dwek, R. A., Burton, D. R., and Prusiner, S. B. (2001) Nature 412, 739-743
132. Heppner, F. L., Musahl, C., Arrighi, I., Klein, M. A., Rulicke, T., Oesch, B., Zinkernagel, R. M., Kalinke, U., and Aguzzi, A. (2001) Science 294, 178-182
133. White, A. R., Enever, P., Tayebi, M., Mushens, R., Linehan, J., Brandner, S., Anstee, D., Collinge, J., and Hawke, S. (2003) Nature 422, 80-83
134. Heppner, F. L., and Aguzzi, A. (2004) Curr Opin Immunol 16, 594-598
135. Korth, C., Stierli, B., Streit, P., Moser, M., Schaller, O., Fischer, R., Schulz-Schaeffer, W., Kretzschmar, H., Raeber, A., Braun, U., Ehrensperger, F., Hornemann, S., Glockshuber, R., Riek, R., Billeter, M., Wüthrich, K., and Oesch, B. (1997) Nature 390, 74-77
136. Moroncini, G., Mangieri, M., Morbin, M., Mazzoleni, G., Ghetti, B., Gabrielli, A., Williamson, R. A., Giaccone, G., and Tagliavini, F. (2006) Neurobiol Dis 23, 717-724
137. Aguzzi, A., Heikenwalder, M., and Polymenidou, M. (2007) Nat Rev Mol Cell Biol 8, 552-561
138. Pilon, J., Loiacono, C., Okeson, D., Lund, S., Vercauteren, K., Rhyan, J., and Miller, L. (2007) Neurosci Lett 429, 161-164
139. Falsig, J., Nilsson, K. P., Knowles, T. P., and Aguzzi, A. (2008) HFSP J 2, 332-341
140. Weissmann, C., and Flechsig, E. (2003) Br Med Bull 66, 43-60
141. Aguzzi, A., Baumann, F., and Bremer, J. (2008) Annu Rev Neurosci 31, 439-477
142. Baumann, F., Tolnay, M., Brabeck, C., Pahnke, J., Kloz, U., Niemann, H. H., Heikenwalder, M., Rulicke, T., Burkle, A., and Aguzzi, A. (2007) EMBO J 26, 538-547
143. Shmerling, D., Hegyi, I., Fischer, M., Blattler, T., Brandner, S., Gotz, J., Rulicke, T., Flechsig, E., Cozzio, A., von Mering, C., Hangartner, C., Aguzzi, A., and Weissmann, C. (1998) Cell 93, 203-214
144. Li, A., Christensen, H. M., Stewart, L. R., Roth, K. A., Chiesa, R., and Harris, D. A. (2007) EMBO J 26, 548-558
145. Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., Steele, A. D., Toyka, K. V., Nave, K. A., Weis, J., and Aguzzi, A. (2010) Nat Neurosci 13, 310-318
146. Clouscard, C., Beaudry, P., Elsen, J. M., Milan, D., Dussaucy, M., Bounneau, C., Schelcher, F., Chatelain, J., Launay, J. M., and Laplanche, J. L. (1995) J Gen Virol 76 ( Pt 8), 2097-2101
147. Everest, S. J., Thorne, L., Barnicle, D. A., Edwards, J. C., Elliott, H., Jackman, R., and Hope, J. (2006) J Gen Virol 87, 471-477
148. Hamir, A. N., Gidlewski, T., Spraker, T. R., Miller, J. M., Creekmore, L., Crocheck, M., Cline, T., and O'Rourke, K. I. (2006) J Vet Diagn Invest 18, 110-114
149. Johnson, C., Johnson, J., Vanderloo, J. P., Keane, D., Aiken, J. M., and McKenzie, D. (2006) J Gen Virol 87, 2109-2114
150. Bruce, M. E. (1993) Br Med Bull 49, 822-838
151. Moore, R. A., Vorberg, I., and Priola, S. A. (2005) Arch Virol Suppl, 187-202
152. Wiltzius, J. J., Landau, M., Nelson, R., Sawaya, M. R., Apostol, M. I., Goldschmidt, L., Soriaga, A. B., Cascio, D., Rajashankar, K., and Eisenberg, D. (2009) Nat Struct Mol Biol 16, 973-978
153. Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova, M. I., Sievers, S. A., Apostol, M. I., Thompson, M. J., Balbirnie, M., Wiltzius, J. J., McFarlane, H. T., Madsen, A. O., Riekel, C., and Eisenberg, D. (2007) Nature 447, 453-457
154. Bessen, R. A., and Marsh, R. F. (1992) J Virol 66, 2096-2101
155. Polymenidou, M., Stoeck, K., Glatzel, M., Vey, M., Bellon, A., and Aguzzi, A. (2005) Lancet Neurol 4, 805-814
156. Peretz, D., Williamson, R. A., Legname, G., Matsunaga, Y., Vergara, J., Burton, D. R., DeArmond, S. J., Prusiner, S. B., and Scott, M. R. (2002) Neuron 34, 921-932
157. Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., Cohen, F. E., and Prusiner, S. B. (1998) Nat Med 4, 1157-1165
158. Herland, A., Nilsson, K. P., Olsson, J. D., Hammarstrom, P., Konradsson, P., and Inganas, O. (2005) J Am Chem Soc 127, 2317-2323
159. Nilsson, K. P., Herland, A., Hammarstrom, P., and Inganas, O. (2005) Biochemistry 44, 3718-3724
160. Nilsson, K. P., Hammarstrom, P., Ahlgren, F., Herland, A., Schnell, E. A., Lindgren, M., Westermark, G. T., and Inganas, O. (2006) Chembiochem 7, 1096-1104
161. Aslund, A., Nilsson, K. P., and Konradsson, P. (2009) J Chem Biol 2, 161-175
162. Sigurdson, C. J., Nilsson, K. P., Hornemann, S., Manco, G., Polymenidou, M., Schwarz, P., Leclerc, M., Hammarstrom, P., Wüthrich, K., and Aguzzi, A. (2007) Nat Methods 4, 1023-1030
163. Behrens, A., and Aguzzi, A. (2002) Trends Neurosci 25, 150-154.
164. Tobler, I., Gaus, S. E., Deboer, T., Achermann, P., Fischer, M., Rulicke, T., Moser, M., Oesch, B., McBride, P. A., and Manson, J. C. (1996) Nature 380, 639-642
165. Manson, J. C., Clarke, A. R., Hooper, M. L., Aitchison, L., McConnell, I., and Hope, J. (1994) Mol Neurobiol 8, 121-127
166. Moore, R. C., Lee, I. Y., Silverman, G. L., Harrison, P. M., Strome, R., Heinrich, C., Karunaratne, A., Pasternak, S. H., Chishti, M. A., Liang, Y., Mastrangelo, P., Wang, K., Smit, A. F., Katamine, S., Carlson, G. A., Cohen, F. E., Prusiner, S. B., Melton, D. W., Tremblay, P., Hood, L. E., and Westaway, D. (1999) J Mol Biol 292, 797-817
167. Rossi, D., Cozzio, A., Flechsig, E., Klein, M. A., Rulicke, T., Aguzzi, A., and Weissmann, C. (2001) EMBO J 20, 694-702
168. Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., Nakatani, A., Kataoka, Y., Houtani, T., Shirabe, S., Okada, H., Hasegawa, S., Miyamoto, T., and Noda, T. (1996) Nature 380, 528-531
169. Mo, H., Moore, R. C., Cohen, F. E., Westaway, D., Prusiner, S. B., Wright, P. E., and Dyson, H. J. (2001) Proc Natl Acad Sci U S A 98, 2352-2357
170. Lührs, T., Riek, R., Güntert, P., and Wüthrich, K. (2003) J Mol Biol 326, 1549-1557
171. Li, A., Sakaguchi, S., Shigematsu, K., Atarashi, R., Roy, B. C., Nakaoke, R., Arima, K., Okimura, N., Kopacek, J., and Katamine, S. (2000) Am J Pathol 157, 1447-1452
172. Behrens, A., and Aguzzi, A. (2002) Trends Neurosci 25, 150-154
173. Behrens, A., Genoud, N., Naumann, H., Rulicke, T., Janett, F., Heppner, F. L., Ledermann, B., and Aguzzi, A. (2002) EMBO J 21, 3652-3658
174. Genoud, N., Behrens, A., Miele, G., Robay, D., Heppner, F. L., Freigang, S., and Aguzzi, A. (2004) Proc Natl Acad Sci U S A 101, 4198-4203
175. Premzl, M., Sangiorgio, L., Strumbo, B., Marshall Graves, J. A., Simonic, T., and Gready, J. E. (2003) Gene 314, 89-102
176. Premzl, M., Gready, J. E., Jermiin, L. S., Simonic, T., and Marshall Graves, J. A. (2004) Mol Biol Evol 21, 2210-2231
177. Uboldi, C., Paulis, M., Guidi, E., Bertoni, A., Meo, G. P., Perucatti, A., Iannuzzi, L., Raimondi, E., Brunner, R. M., Eggen, A., and Ferretti, L. (2006) Mamm Genome 17, 1130-1139
178. Watts, J. C., Drisaldi, B., Ng, V., Yang, J., Strome, B., Horne, P., Sy, M. S., Yoong, L., Young, R., Mastrangelo, P., Bergeron, C., Fraser, P. E., Carlson, G. A., Mount, H. T., Schmitt-Ulms, G., and Westaway, D. (2007) EMBO J 26, 4038-4050
179. Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C., Lopez Garcia, F., Billeter, M., Calzolai, L., Wider, G., and Wüthrich, K. (2000) Proc Natl Acad Sci U S A 97, 145-150
180. Koradi, R., Billeter, M., and Wüthrich, K. (1996) J Mol Graphics 14, 51-57
Contributed by
Aguzzi, A. and Hornemann, S., including contributions to the Eighth ICTV Report by Prusiner, S.B., Baron, H., Carlson, G., Cohen, F.E., DeArmond, S.J., Gabizon, R., Gambetti, P., Hope, J., Kitamoto, T., Kretzschmar, H.A., Laplanche, J.-L., Tateishi, J., Telling, G. and Will, R.
Figures
Figure 1 Organization and structural properties of the mouse prion protein. (A) Scheme of the organization of the mouse prion protein (numeration according to the human prion protein). In the mature form of the protein the N-terminal signal peptide (N-terminal green box) is cleaved off and the C-terminal peptide (C-terminal green box) is replaced by a GPI anchor at Ser-231. The N-terminal unstructured part of the protein contains five octarepeats (magenta boxes), two positively charged amino acid clusters CC1 and CC2 (orange boxes) and a highly conserved hydrophobic polypeptide segment comprising residues 111134 (yellow box). The folded C-terminal domain contains three -helices H1H3 (blue boxes) and a short antiparallel -sheet (indicated by two black arrows). Helices 2 and 3 are connected by a single disulfide bridge between Cys-179 and Cys-214. The protein also contains two potential glycosylation sites at amino acid positions 181 and 197 (light blue ellipses). (B) Three-dimensional structure of mPrP(121231). The three -helices are shown in blue and the antiparallel -sheet in green. (C) Electron micrographs of negatively stained recombinant mouse fibrils. The bar represents 100 nm.
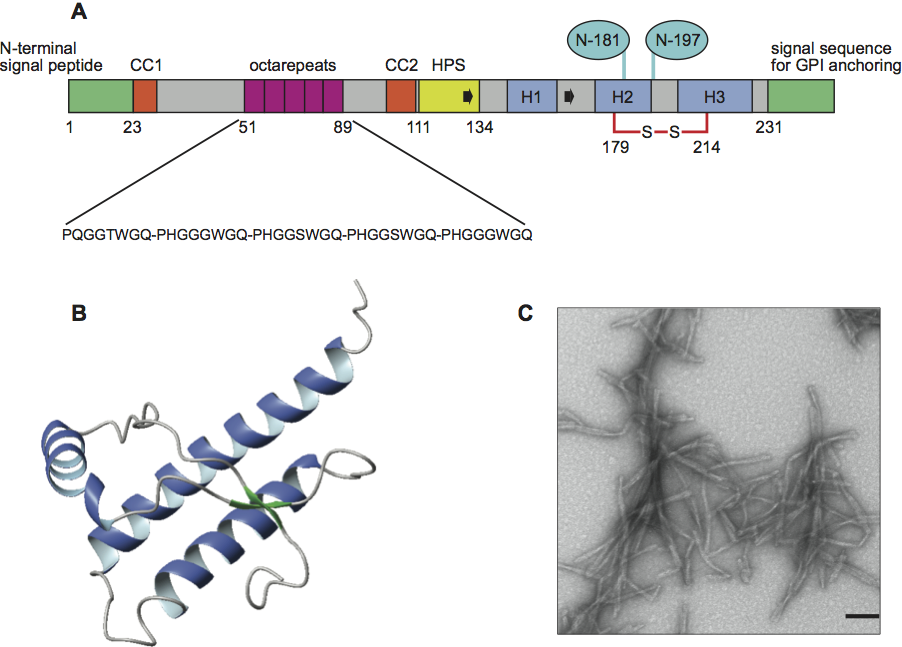
Figure 2 Location of point mutations in the three-dimensional structure of hPrP(121230) that are associated with familial prion diseases. The backbone is shown in grey and the side chains of the residues associated with the mutations for inherited fCJD are shown in red (A), for GSS in blue (B) and fFFI in magenta (C). The polymorphism at position 129 is indicated in green. The molecules were generated with the program MOLMOL.
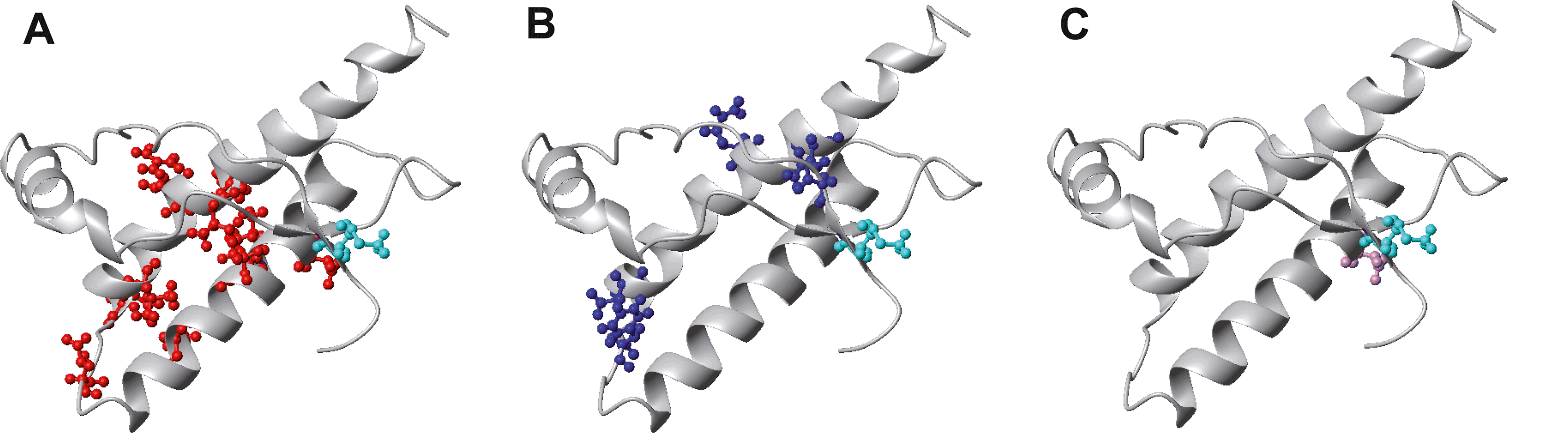
Figure 3 Comparison of images showing different prion strains stained with PTAA and Thioflavin T. (A) MDePrPSc, (B) OvPrPSc and (C) BoPrPSc prion deposits. PTAA-stained deposits appear in yellow-red. Scale bars represent 500 m.
(Adapted from Sigurdson, C.J., Nilsson, K.P., Hornemann, S. et al. (2009). Proc. Natl Acad. Sci., U S A, 106, 304309.)