


9th – 11th June, 2016
Simmons College, Harvard Campus, Boston, USA
Sponsored by the Wellcome Trust
Organized by members of the ICTV Executive Committee
A workshop funded by the Wellcome Trust (UK) to discuss frameworks for advancing viral taxonomy in the age of metagenomics was convened in Boston, MA, USA from 9-11 June 2016. It was organized and chaired by Peter Simmonds and administered locally by Max Nibert. Participants had wide-ranging expertise in viral genomics, metagenomic environmental studies, and virus classification (13 of the 26 participants were members of the International Committee on Taxonomy of Viruses (ICTV) Executive Committee) and based on data presentations and wide-ranging discussions set out to develop a series of expert proposals for future consideration by the ICTV Executive Committee.
The understanding in the workshop was that the term metagenomic applies to any viral sequence that lacks biological or other experimental characterization, although what a ‘lack’ means in practice has varied in the literature. Sequence data are already of paramount importance in viral taxonomy because they currently provide the only reliable means of representing evolutionary relationships at the required granularity; however, the workshop recognized that the data generated by high-throughput sequencing from environmental samples pose major challenges, particularly because increasingly powerful methods are generating overwhelming amounts of such data, which are linked to little or no biological information.
The workshop participants concluded that it is entirely valid to use metagenomic sequences in virus taxonomy in the absence of an isolate or direct biological data, such as visualization of viral particles or detection of signs or symptoms of disease. A set of proposals was developed and are discussed in the consensus Statement article published in Nature Reviews Microbiology. These proposals were subsequently endorsed by the ICTV Executive Committee.
Mya Breitbart
University of South Florida, College of Marine Science, 140 7th Avenue South
Saint Petersburg, FL 33701, USA
Rodney Brister
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA
Eric B. Carstens
Department of Microbiology and Immunology, Queen's University, Kingston, ON K7L 3N6, Canada
Eric Delwart
Blood Systems Research Institute, Department of Laboratory Medicine, University of California San Francisco, San Francisco, CA, 94118, USA
Roger Hull
Formerly John Innes Centre, Norwich Research Park, NR4 7UH Colney, Norwich, UK
Eugene V. Koonin
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, 20894, USA
Marilyn J. Roossinck
Department of Plant Pathology and Environmental Microbiology, Center for Infectious Disease Dynamics, Pennsylvania State University, University Park, PA 16802, USA
Matthew B. Sullivan
Soil, Water, and Environmental Science, University of Arizona, Tucson, AZ 85721, USA
Curtis A. Suttle
Departments of Earth, Ocean & Atmospheric Sciences, Microbiology & Immunology, and Botany, University of British Columbia, Vancouver, V6T 1Z4, Canada & Canadian Institute for Advanced Research, 180 Dundas St W, Toronto ON, M5G 1Z8, Canada
Robert B. Tesh
Department of Pathology and Center for Biodefense and Emerging Infectious Diseases, University of Texas Medical Branch, Galveston, TX 77555-0609, USA
Mike J. Adams
24 Woodland Way, Stevenage, Herts, SG2 8BT, UK
Andrew J. Davison
MRC-University of Glasgow Centre for Virus Research, Sir Michael Stoker Building, 464 Bearsden Road, Glasgow, G61 1QH, UK
Alexander E. Gorbalenya
Department of Medical Microbiology, Leiden University Medical Center, P. O. Box 9600, E4-P, rm. E4-72, 2300 RC, Leiden, The Netherlands
Balázs Harrach
Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences, 21 Hungária krt., Budapest, H-1143, Hungary
Andrew M.Q. King
Sunfield, Dawney Hill, Pirbright, Woking, Surrey, GU24 0JB, UK
Mart Krupovic
Department of Microbiology, Institut Pasteur, 25 rue du Dr Roux, 75015, Paris, France
Jens H. Kuhn
NIH/NIAID/DCR Integrated Research Facility at Fort Detrick (IRF-Frederick), B-8200 Research Plaza, Fort Detrick, Frederick, MD, 21702, USA
Elliot J. Lefkowitz
Department of Microbiology, University of Alabama at Birmingham (UAB), BBRB 276, 845 19th ST South, Birmingham, AL, 35294-2170, USA
Max Nibert
Department of Microbiology and Immunobiology, Harvard Medical School, 77 Ave Louis Pasteur, Boston, MA, 02115, USA
Sead Sabanadzovic
Department of Biochemistry, Molecular Biology, Entomology and Plant Pathology, Mississippi State University, 100 Old Hwy 12 Mail Stop 9775, Mississippi State, MS, 39762, USA
Peter Simmonds
Nuffield Department of Medicine, University of Oxford, South Parks Road, Oxford, OX1 3SY, UK
Arvind Varsani
The Center for Fundamental and Applied Microbiomics, The Biodesign Institute and School of Life Sciences, Arizona State University, Tempe, AZ, USA
Murilo Zerbini
Dep. de Fitopatologia/BIOAGRO, Universidade Federal de Viçosa, Viçosa, MG, 36570-900, Brazil
Mariá Benkő
Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences, 21 Hungária krt., Budapest, H-1143, Hungary
René A. van der Vlugt
Plant Research International, Wageningen University and Research Centre, Droevendaalsesteeg 1, 6708 PB, Wageningen, The Netherlands
Richard Orton
MRC-University of Glasgow Centre for Virus Research, Sir Michael Stoker Building, 464 Bearsden Road, Glasgow, G61 1QH, UK
9th – 11th June, 2016
Simmons College, Harvard Campus, Boston, USA
The presentations and discussions will take place over 5 half day sessions (Thursday, am, pm, Friday, am, pm and Saturday, am).
Meetings will start at 8.45 on the three days. On Thursday and Friday, lunch will be available between 12.30 – 1.45 and the meeting will finish by 6.00 at the latest. On Saturday, the meeting will run from 8.45 and conclude at 12.00.
A) Introduction and Overview. An outline of the aims and purpose of the meeting, focussed discussions and planned outputs
Introduction to Harvard: Max Nibert
Aims and plans for the meeting: Peter Simmonds
The process of virus classification and the role of the ICTV: Mike Adams, Andrew Davison
How are other organisms classified? How are metagenomic sequences handled elsewhere?: Peter Simmonds
B) Presentations. Identification of the problems with current classification methods, themed presentation from invited experts, 15 minutes, description of specific issues, problems and current pragmatic solutions. While the number of speakers mandates a restriction in the length of each presentations, the time spent on discussion will not be proscribed and indeed will constitute an essential part of the resolution
Vertebrate viruses / arboviruses: Eric Delwart, Bob Tesh
Environmental:Arvind Varsani, Mya Breitbart, Curtis Suttle
Plant:Marilyn Roossinck, Roger Hull
Bacteriophage: Matthew Sullivan, Mart Krupovic
Evolutionary process and methodologies: Eugene Koonin
Database Management: Rodney Brister
C) General Discussion. General discussion session with invited experts and the following EC members participating in the meeting. Identification of common and virus-group-specific classification issues and potential strategies for incorporating metagenomic sequence data into the virus classification:
D) Resolution and meeting outputs
Published as Supplementary information S1 in:
Nature Reviews Microbiology (2017)
doi:10.1038/nrmicro.2016.177
Published online 03 January 2017
The metagenomics working group (MWG) comprised an international group of invited experts from the field of metagenomics and representative members of the ICTV Executive Committee (Meeting attendance is listed in Appendix I). The meeting was host to an extensive discussion of the challenges posed by the abundance of MG sequence data currently being generated, the procedures currently being used by the ICTV to classify viruses and the strategies by which the two could be reconciled.
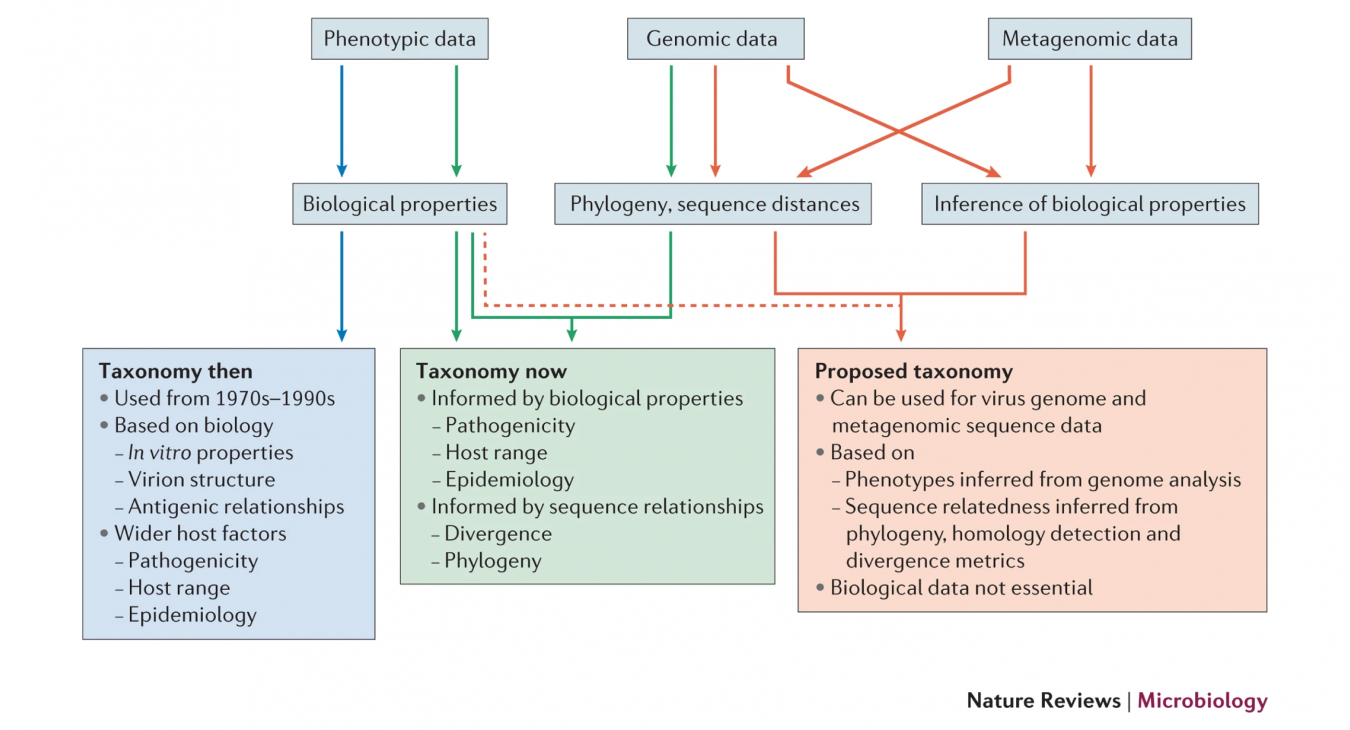
The meaning of the term “Metagenomic”. There was substantial agreement in most areas of the discussion between experts and the ICTV EC. Firstly, the MWG as a whole were in agreement about the meaning of the term “Metagenomic” as applied to viruses, despite the wide range of ways in which this term has been used in the literature and the fact that there is a continuum of variability in the extent of other information available for viruses characterised from environmental sampling. It was additionally recognised that MG sequences are not simply to be equated with those from next generation sequencing methods (NGS), and that viral sequences that frequently lack other biological or experimental information can be equally obtained by PCR and other virus characterisation methods. Nevertheless, it is the unprecedented effectiveness of NGS in characterising viral populations in the environment that has largely created the classification problems we currently face.
There was similar unanimity in the supposition that, with appropriate caveats, detection of a viral sequence in a sample equates to the presence of a virus. It is consequently entirely valid to use such sequences for virus classification in the absence of a virus isolate or other evidence for the physical presence of a virus (such as visualisation of virus particles, disease symptoms in an infected animal or plant, etc.). Furthermore, the MWG considered that, under the appropriate methodological conditions, viral sequences assembled from NGS data were sufficiently accurate and reliable to be used for classification processes.
Technical limitations. The group however acknowledges the potential problems with deriving viral sequences from mixed viral populations in the sample and the consequent danger in certain situations of assembling chimeric genomes. It was also recognised that current methodology often cannot assemble complete genome sequences from viruses with segmented genomes and multipartite genomes packaged into separate virus particles. Particularly for the latter, the means to reliably link together sequences from different genome segments may remain unavailable. Another practical problem arises from the presence of virus-derived sequences into the genomes of the infected host, often in the germline, incapable of generating infectious virus although often transcribed. These are all caveats that must be addressed experimentally for MG sequence data to be used for classification purposes. These are, however, not fundamental barriers to classification as the technology used to create MG sequences is continuously improving and many of the current technical problems, particularly with assembly, will be resolved.
The ICTV species definition. The current definition of species formulated by the ICTV is:
A species is the lowest taxonomic level in the hierarchy approved by the ICTV. A species is a monophyletic group of viruses whose properties can be distinguished from those of other species by multiple criteria
The MWG acknowledged the concern expressed by many virologists that an assembled MG sequence derived from environmental sampling lacks the “multiple criteria” required for a species definition. Such criteria are traditionally information on its biology (host range, epidemiology, disease associations), morphology, and behaviour in cell culture, many of which are used as defining features of different species.
However, it was considered by the MWG that a genome sequence, even without associated other biological information, can possess sufficiently varied attributes to support the creation of a species. In addition to phylogenetic analyses using robust models, these may include:
Collectively, these attributes provide the required information on its monophyly and bioinformatic characterisation of the virus that satisfies the multiple criteria required for a species definition. The sequence furthermore provides considerable further information on its evolutionary history and relationships with other viruses at different taxonomic layers.
Assignment of MG sequence to existing virus families. The MWG recognises the range of ways and criteria by which viruses in the existing classification are assigned to species and genus ranks within the currently designated virus families. These “classification frameworks” are specific to each virus family and are based on combinations of factors that may include both biological information (eg. host range, geographical distribution or pathogenesis) and genetic attributes such a degree of sequence divergence and gene complements or other aspects of genome organisation.
While biological information can be part of the definition of existing taxa, there is invariably associated sequence data for members of each. Thus, while a MG sequence may lack the biological information previously by ICTV to support assignment in a particular taxon, assignment of new species and genera to an existing virus family with MG sequences is permissible if sequence relationships (phylogeny, sequence divergence, gene complements) are equivalent to those that exist among viruses classified by other means. Assignment of MG viruses as new species is possible within existing ICTV rules and has already been done in a number of families in recent years..
Classification of MG sequences into new families. The MWG recognises that the assignment of MG sequence as members of newly created families is subject to different constraints and difficulties to the previously discussed addition of MG sequences an existing virus family with a classification framework. This is because criteria used for an informative subdivision of viruses into different genera and species vary so greatly between existing virus families and cannot be reliably predicted a priori in the absence of considerable information of virus relationships within the family. Particularly for families of viruses infecting animals and plants, species definitions are often defined by disease or other biological attributes. While these divisions are informative, there is consequently huge variability in the degree of sequence divergence between species and genera that cannot be predicted from inspection of sequence data alone.
For creation and assignment of MG sequences to new virus families, the absence of biological information requires that taxon levels must be governed by clustering and patterns of variability between MG sequences. This requires a considerable amount of comparative sequence information for viruses to be assigned to the family for the assignment of genus and species levels to be informative and to possess practical utility.
The MWG unanimously decided that new virus families populated entirely by viruses identified from MG sequence data could be created. There was, however, substantial agreement on the caveat that multiple examples of members of the new virus family would be required to establish a classification framework for lower taxa. Such variants would provide the required information on genetic divergence and similarities in genome organisation and gene complements for designation of appropriate genus and species categories.
MG sequence nomenclature. Most of the MWG considered that the ICTV should not be proscriptive about the formation on names and that practices currently used within individual Study Groups (SGs) for taxon nomenclature could be applied equally to new taxa created for MG sequences. This recognises the fact established after some discussion that it is the component viruses within a taxon that may or may not be metagenomically derived, not the taxon itself. It is quite conceivable that a taxon may contain viruses derived by multiple methods. Furthermore, a species comprising MG sequences only may ultimately come to contain an additional member derived from virus isolation and with defined biological properties. Thus MG status belongs to and is recoverable from the sequence record, not from the taxon it is assigned to.
MG taxon proposals (TPs). The MWG was in substantial agreement that the TP procedure could be improved though electronic submission and appropriate quality checks and this is indeed under development by the ICTV. TPs allow multiple new species to be proposed in the same submission form which is of value for classification of large MG datasets.
The MWG were largely in agreement that with appropriate oversight and under appropriate circumstances, species assignments could be made at the discretion of the relevant SG and sub-Committee chair without the degree of scrutiny normally required by the EC. This would allow species designations to incorporated into the ICTV classification on a continuous basis and much more rapidly that the current annual procedure. This would be a benefit to the virology community and to the public databases which seek to provide classification information on new sequence submissions. There would be some procedural difficulties associated with this change but the EC will discuss the possibility.
There was considerable discussion on the possibility of creating more higher level taxonomic groupings, such as orders, that would recognise relatively distant relationships between existing virus families and also provide a placeholder for sequence databases and Study Groups to use for otherwise unclassified MG sequences. Typically the latter may include viruses without close relatives and for which a formal designation into family, genus and species might be inappropriate or premature in the absence of a classification framework with which to usefully specify those taxonomic layers.. A much discussed example was a potential new Order containing viruses with ssDNA circular genomes and a Rep gene as there are large numbers of such sequences that can only currently be described as unclassified viruses.
Attendees of the Workshop, Metagenomic sequencing and its impact on virus classification, published an article summarizing the workshop discussions as a Consensus Statement in Nature Reviews Microbiology:
Nature Reviews Microbiology (2017); doi:10.1038/nrmicro.2016.177; Published online 03 January 2017
A comment on this article has been published by the Editors of Nature Reviews Microbiology in: "A sea change for virology", Nature Reviews Microbiology 15, 129 (2017).
{kind=link}